Effective clinical trial document lifecycle management encompasses the entire history of a document, from its initial creation through archival. This is not an administrative filing task but a systematic, controlled process for managing essential documents like protocols, investigator's brochures, and clinical study reports. The objective is to ensure these documents are current, compliant, and continuously ready for regulatory inspection. This process is a core strategic function for sponsors, CROs, and clinical sites.
The Strategic Core of Inspection Readiness
Robust document management is the foundation of a trial's integrity and compliance. It ensures that every piece of documentation, from the protocol to the final Clinical Study Report (CSR), tells a clear and defensible story of the trial's conduct. Without a structured lifecycle, this narrative can become fragmented, creating vulnerabilities during regulatory inspections.
A useful analogy is to view a clinical trial as a house under construction. The protocol is the blueprint, the investigator's brochure details the materials, and the Trial Master File (TMF) serves as the complete record of how the house was built. Mismanagement of any document introduces structural flaws. An outdated informed consent form is analogous to a compromised foundation. A protocol with inadequate version control creates weaknesses in the structure. Either scenario jeopardizes the integrity of the entire project.
High Stakes and Proactive Management
The consequences of inadequate document management are significant. Regulatory authorities often interpret documentation gaps as a lack of operational control. This can lead to costly submission delays due to misaligned regional requirements or missing information. According to CCRPS.org, documentation issues are a primary contributor to delays in trial startup timelines.
Proactive lifecycle management transforms documentation from a reactive administrative burden into a strategic asset. For sponsors and CROs, it is essential for mitigating regulatory risk and demonstrating the credibility of trial data.
A well-managed lifecycle creates a clear, auditable trail that demonstrates operational control and foresight. Each stage—authoring, review, approval, distribution, and archival—functions as a critical quality checkpoint. This structured approach is fundamental to building a robust TMF that is inspection-ready at all times. For further detail on TMF structure, the DIA Trial Master File Reference Model provides a comprehensive framework.
Ultimately, this discipline is not about achieving administrative perfection. It is about protecting patient safety, maintaining data integrity, and communicating the scientific and ethical narrative of the trial with absolute clarity. Mastering the document lifecycle is fundamental to successful clinical development.
A Document's Journey: The Five Stages of Its Lifecycle
To effectively manage clinical trial documentation, it is essential to view each document as having a lifecycle with a distinct beginning, middle, and end. Every document, from a simple log to a complex protocol, follows a predictable path. Adherence to this path is key to maintaining a compliant, organized, and inspection-ready trial.
Let's examine this journey using one of the most critical documents: the Clinical Study Protocol (CSP). The protocol serves as the master blueprint for the entire trial. If its lifecycle is poorly managed, the entire trial's structural integrity can be compromised.
Stage 1: Authoring – Laying the Foundation
The process begins with the translation of a scientific concept into a structured document. The authoring stage involves more than just writing; it is about constructing a document that is scientifically sound, operationally feasible, and aligned with regulatory expectations from the first draft.
For a protocol, this typically involves starting with a template aligned with guidelines such as ICH E6. This operational best practice helps ensure all required sections are included. Medical writers and clinical scientists then collaborate to populate the template, detailing elements from study objectives and patient selection to statistical methods and safety monitoring plans.
The primary goal at this stage is precision. Any ambiguity introduced during authoring can lead to confusion, protocol deviations, and regulatory queries later in the process.
Stage 2: Review – The "Many Eyes" Principle
Once a draft is complete, it enters the review stage. This is a collaborative phase where subject matter experts from various functions pressure-test the document to ensure its viability from multiple perspectives.
A parallel review process, where different functional areas provide feedback simultaneously, is an efficient operational practice:
- Medical Team: Verifies scientific and clinical accuracy, assessing the adequacy of patient safety measures and the validity of study objectives.
- Biostatistics: Scrutinizes the study design, sample size calculations, and the statistical analysis plan to confirm the protocol can generate the data required to answer the research question.
- Clinical Operations: Evaluates the protocol from a practical, real-world perspective. This team assesses whether site staff can realistically execute the procedures and whether the burden on participants is acceptable.
- Regulatory Affairs: Ensures the protocol complies with the specific regulations of every country where the trial will be conducted.
This collaborative review is designed to identify and resolve potential issues before they become costly, study-delaying problems. All comments, suggestions, and their resolutions become part of the document's permanent audit trail.
Stage 3: Approval – The Official Green Light
After all feedback has been incorporated and consensus is reached, the document proceeds to final approval. This is the formal sign-off that transitions a draft into an official, effective document.
Systems used for this stage must comply with regulations governing electronic records and signatures, such as FDA 21 CFR Part 11. These regulations provide the framework for ensuring electronic signatures are as legally binding as handwritten signatures.
An electronic signature is more than a digital image; it is a secure, time-stamped, and traceable record that provides verifiable proof of who approved a document and when. It is a cornerstone of a compliant audit trail.
Once final signatures are applied, the document is locked, finalized, and assigned an official version number (e.g., "Protocol Version 1.0"). From this point forward, it is the single source of truth. Any subsequent modifications will require a formal amendment, which initiates a new lifecycle for that version.
Stage 4: Distribution – Getting It into the Right Hands
An approved protocol must be distributed to the teams who will execute it, including investigator sites, CROs, and internal personnel. This distribution must be a controlled process. Distributing an incorrect version to a site poses a significant compliance and patient safety risk.
A controlled distribution process ensures that:
- The final, approved version is sent to a specific, pre-defined distribution list.
- Receipt and acknowledgment of training on the document can be tracked.
- All stakeholders know where to access the current effective version, and superseded versions are clearly identified or removed from general access.
This stage is about ensuring operational consistency. When every site operates from the same set of instructions, the result is reliable data and, most importantly, the protection of patient safety.
Stage 5: Archival – Preserving the Record
The document lifecycle does not end when the trial concludes or a new version is issued. The final stage is archival. Regulations require that trial documents be stored securely for extended periods—sometimes 25 years or more, depending on the product and jurisdiction.
This involves transferring the final document and its complete history (including drafts, review comments, signatures, and distribution records) into the electronic Trial Master File (eTMF). The eTMF is the official repository for the trial, organized to provide a complete narrative for an auditor.
Proper archival ensures the document remains secure, readable, and accessible for decades. It closes the lifecycle loop, creating a permanent, immutable record that serves as evidence of a well-conducted, compliant clinical trial.
Understanding the Regulatory Frameworks for Document Management
Managing clinical trial documents is not just a matter of organizational preference; it is a requirement dictated by a global network of regulations. These frameworks provide the rules for presenting a verifiable account of a trial's conduct to regulatory authorities. Their primary purpose is to protect trial participants and ensure the integrity of the collected data.
The foundational guideline is ICH E6(R3) Good Clinical Practice (GCP). This harmonized global standard outlines the roles and responsibilities of all parties involved, from the sponsor to the investigator. GCP principles require rigorous trial oversight, which can only be demonstrated through complete and contemporaneous documentation. Every document, from the initial protocol to the final site training log, serves as evidence that the trial was conducted ethically and scientifically.
Core Guidelines That Shape How We Write
Beyond the overarching principles of GCP, specific guidelines dictate the structure and content of critical documents. For example, ICH E3 provides a detailed template for the Clinical Study Report (CSR). Following this structure is not a box-ticking exercise; it ensures that regulators receive a consistent, logical summary of the trial, facilitating their review for a potential new drug approval.
This principle applies to other key documents as well. The Statistical Analysis Plan (SAP), for instance, is based on the principles outlined in ICH E9. These standards represent an industry consensus on how to present complex trial data with clarity, reducing ambiguity so that health authorities can efficiently assess the findings.
The process a document follows to meet these standards involves multiple steps, moving from a draft to a final, controlled record.
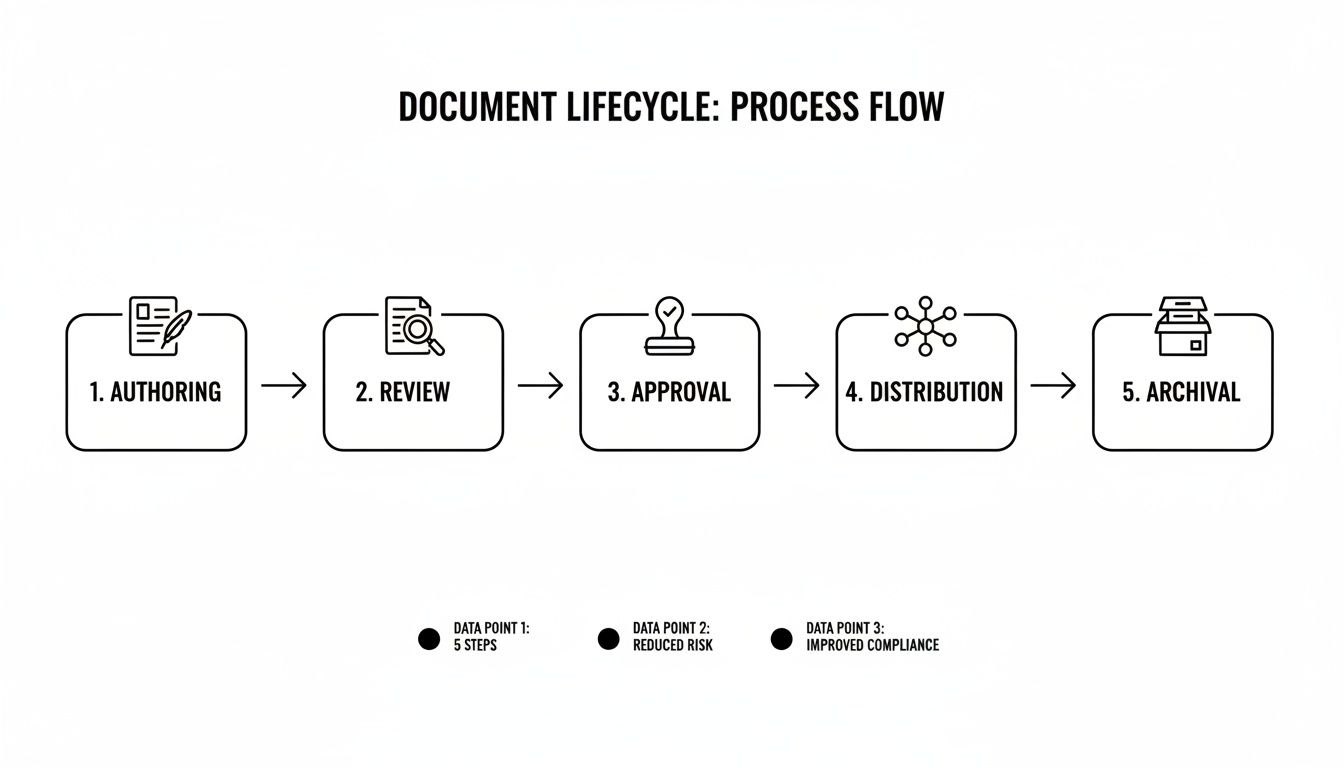
This flow illustrates that each stage is a critical quality checkpoint. The intent is to build quality and compliance into the document from its inception, long before it is reviewed by an auditor.
The Role of National and Regional Regulations
While ICH provides a global framework, national and regional regulations add legally binding requirements. In the United States, the FDA’s 21 CFR Part 11 sets the standard for electronic records and signatures, defining the criteria that make them trustworthy and reliable. This regulation directly impacts the selection and validation of software used for document management, which must include features like secure access controls, time-stamped audit trails, and validated electronic signatures. A detailed examination of this topic can be found in our guide on achieving FDA 21 CFR Part 11 compliance.
In Europe, the EU Clinical Trials Regulation (EU CTR 536/2014) has fundamentally altered the regulatory landscape by introducing the Clinical Trial Information System (CTIS). This system creates a single, harmonized submission portal for all EU member states, centralizing documentation and enhancing the transparency of the entire trial lifecycle.
These regulations work in concert to create a system of checks and balances. Their goal is to ensure the final data is Attributable, Legible, Contemporaneous, Original, and Accurate (ALCOA)—the five pillars of data integrity.
Technology and regulatory harmonization are driving a significant shift in clinical operations. Recent analyses indicate a substantial increase in trial initiations, particularly in the decentralized trial model, which demands even more robust digital systems. However, underreporting of trial initiations remains an issue, with an estimated 13% gap due to disclosure delays. This highlights the critical importance of compliant, real-time documentation from the outset. As noted by Clinical Trials Arena, the full implementation of the EU CTR is a key driver of this transformation.
These regulatory frameworks are not merely bureaucratic hurdles. They are essential guardrails that ensure every aspect of a clinical trial is meticulously documented, auditable, and defensible, protecting both the trial participants and the future patients who will rely on the trial's outcome.
Defining Key Roles and Responsibilities in Document Workflows
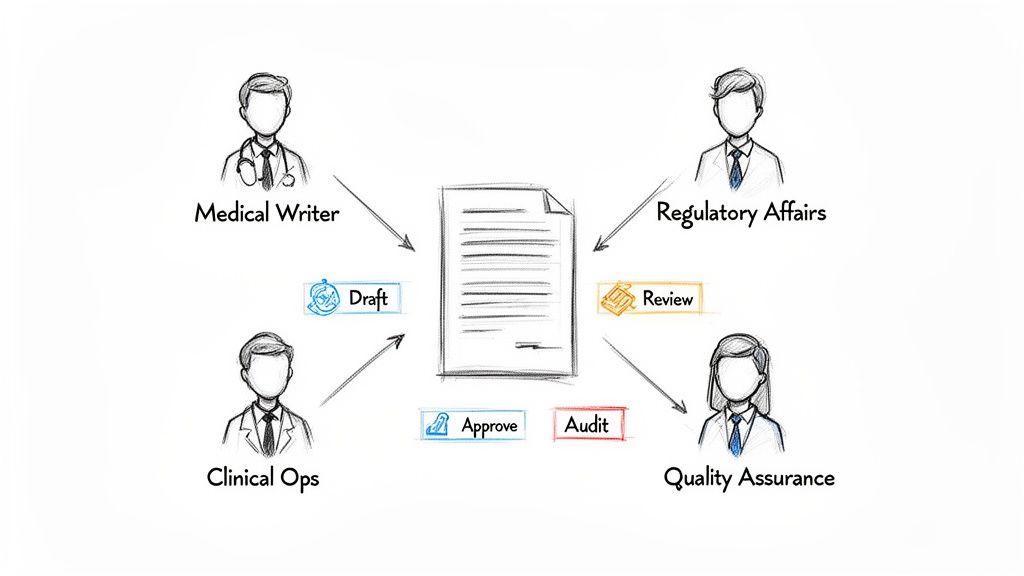
A robust process for managing clinical trial documents requires clearly defined roles and responsibilities. Without this clarity, document workflows can stall, creating operational bottlenecks that may jeopardize timelines and introduce compliance risks.
The process can be compared to a relay race, where each team member has a specific function, and handoffs must be seamless. Each role contributes a different expertise, ensuring that documents are not only scientifically rigorous but also operationally feasible for clinical sites and prepared for regulatory review.
Core Players in the Document Lifecycle
While specific job titles may vary between organizations, the core functions involved in the document lifecycle are consistent across the industry.
-
Medical Writers: These individuals are responsible for authoring the document. They translate complex scientific and clinical data into a clear, compliant narrative for documents such as protocols, clinical study reports (CSRs), and Investigator's Brochures. They typically create the initial draft.
-
Regulatory Affairs Specialists: This team serves as the liaison with health authorities. As compliance experts, they review documents to ensure they meet the stringent requirements of agencies like the FDA and EMA, verifying that both content and format are submission-ready.
-
Clinical Operations Managers: This function provides the perspective of the clinical site. They conduct an essential real-world review to confirm that documents are operationally sound. They address practical questions, such as, "Can a site reasonably perform these procedures?" or "Is this informed consent form sufficiently clear for a patient?"
-
Quality Assurance (QA) Professionals: QA acts as an independent oversight function. They audit the final document and, equally important, the process used to create it. Their role is to verify that all activities were conducted in accordance with internal SOPs and GCP principles, ensuring a defensible audit trail.
Putting It All Together: An Investigator's Brochure Update
Consider a common scenario: updating an Investigator's Brochure (IB) in response to new safety data.
-
Drafting: The Medical Writer initiates the process by synthesizing the new nonclinical and clinical data and drafting the updated sections of the IB, focusing on scientific precision and clarity.
-
Compliance Review: The draft is then reviewed by Regulatory Affairs. This team examines the changes against global standards to ensure the new information is presented in compliance with the requirements of all relevant health authorities.
-
Final Audit: Before final approval, the updated IB and its complete change history are reviewed by Quality Assurance. QA confirms that the update process adhered to SOPs, all reviews were properly documented, and the final version is suitable for distribution.
A responsibility assignment matrix, commonly known as a RACI chart, is an effective tool for formalizing these interactions. It clarifies who is Responsible for executing the work, who is Accountable for the final outcome, who must be Consulted for input, and who needs to be kept Informed.
The following is a simplified RACI matrix for a common task: amending a clinical trial protocol.
Responsibility Matrix for a Protocol Amendment
| Task | Medical Writer | Clinical Operations | Statistician | Regulatory Affairs | Quality Assurance |
|---|---|---|---|---|---|
| Drafting the amendment text | R | C | C | I | I |
| Reviewing for operational feasibility | I | R | I | C | I |
| Validating statistical plan changes | C | I | R | I | I |
| Ensuring regulatory compliance | C | C | I | R | I |
| Final Quality Control (QC) check | I | I | I | I | R |
| Final approval and release | I | A | I | A | C |
This level of clarity is operationally valuable. Using a RACI matrix reduces confusion, defines ownership, and makes every action traceable. This structured approach helps build the unambiguous audit trail that regulatory inspectors expect, forming a cornerstone of effective clinical trial document lifecycle management.
Using Technology to Bolster Lifecycle Management
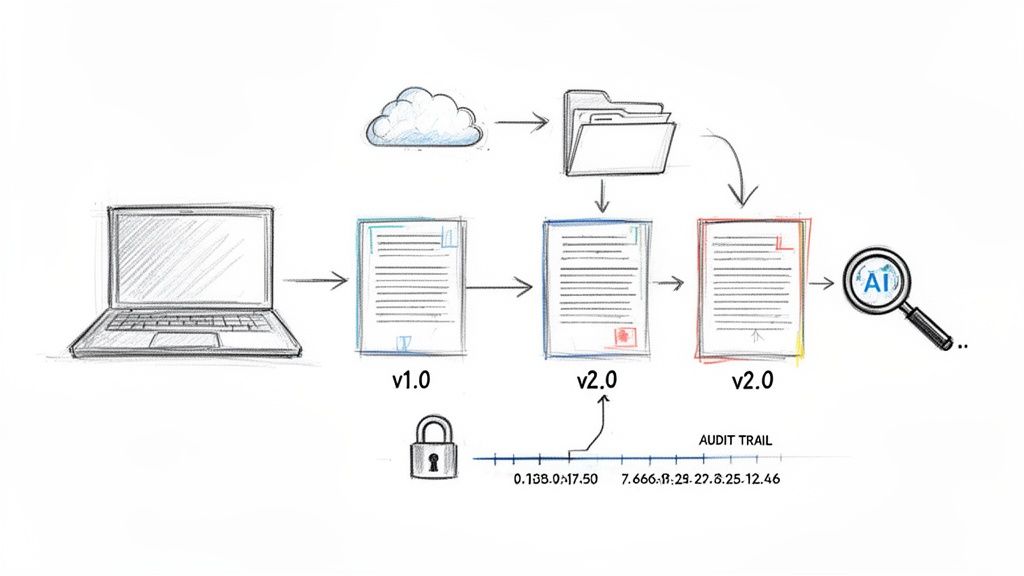
In modern clinical trials, technology is the backbone of effective clinical trial document lifecycle management. It provides the means to translate regulatory theory into practical, real-world application. These systems are not merely digital storage repositories; they are controlled environments designed to enforce compliance, provide clear oversight, and protect the integrity of every document from creation to archival.
At its core, this technology automates processes that are susceptible to human error. By embedding standard operating procedures (SOPs) directly into digital workflows, these systems ensure that established rules are followed consistently. This is critical for demonstrating operational control during a regulatory inspection.
Automating Control and Ensuring Version Integrity
A primary challenge that technology addresses is version control. Ensuring that every clinical site is using the single correct, currently approved version of a protocol or informed consent form is a matter of patient safety and data quality where there is no margin for error.
Automated systems achieve this through:
- Locking approved documents to prevent unauthorized modifications.
- Managing distribution lists to disseminate new versions to authorized personnel only.
- Superseding outdated versions automatically, removing them from active use to prevent inadvertent use of incorrect information.
This creates a complete, time-stamped audit trail for every document. It is a direct reflection of Good Clinical Practice (GCP) in action, providing a verifiable record of who performed which actions, and when, throughout the document's lifecycle.
Centralizing Documents for Inspection Readiness
Ultimately, all essential trial documents are consolidated in a central repository: the electronic Trial Master File (eTMF). An eTMF is more than a storage system; it is a dynamic library structured to provide a real-time view of the trial's status and to be inspection-ready at all times. A compliant eTMF cannot be maintained without robust lifecycle management for each contributing document.
The purpose of an eTMF is to maintain the trial's story in a constant state of readiness. It allows any authorized party—sponsors, CROs, auditors—to reconstruct events at any point in time to verify that the trial was conducted correctly and that the data is trustworthy.
Integrating authoring, review, and approval workflows directly with the eTMF ensures that final, approved documents are filed correctly and contemporaneously. This eliminates manual, error-prone steps that can create compliance gaps. To explore this topic further, consider our detailed guide on electronic trial master file software.
The Role of AI in Supporting Human Expertise
Emerging technologies like artificial intelligence (AI) are beginning to play a significant role, primarily by supporting human experts rather than replacing them. AI-powered tools are effective for performing large-scale quality control checks, identifying issues that may be tedious or easily missed by human reviewers. For example, an AI can scan thousands of documents to detect inconsistent terminology or missing metadata, allowing clinical professionals to focus on higher-value scientific and operational tasks.
AI is transforming manual QC tasks into highly efficient, inspection-ready workflows. The industry is also moving toward concepts like Digital Data Flow (DDF), where a structured protocol can be used to automatically generate other documents, such as case report forms, thereby reducing manual errors. Some modern eTMF systems can already auto-classify documents and flag quality issues in near real-time. You can learn more about these trends in this insightful 2025 outlook.
Common Questions on Document Lifecycle Management
Professionals in clinical and regulatory roles frequently encounter complexities in managing trial documentation. The following addresses several common questions to clarify key concepts and operational hurdles.
Document Management Versus TMF Management
These two concepts are often confused, but the distinction is straightforward.
Document management pertains to the lifecycle of a single document. For example, it includes the entire process from the initial draft of a study protocol, through all review and revision cycles, to final approval and distribution. It is the history of an individual file.
TMF management, on the other hand, is the overarching process. It involves collecting the final protocol, along with all other essential trial documents, and organizing them into a complete, inspection-ready repository. Effective TMF management is dependent on sound lifecycle management for every document it contains.
How Version Control Functions in a Regulated Environment
In a regulated context, version control is not merely about saving different drafts; it is a critical system for ensuring that all personnel are using the correct and current document. This is non-negotiable for maintaining patient safety and data integrity.
This is a disciplined, step-by-step process:
- Each time a document is finalized and approved, it is assigned a distinct version number, such as Protocol V1.0, followed by V2.0 for the next approved version.
- A complete history of all changes made between versions is tracked and maintained for traceability.
- Once a new version is approved and distributed, the previous version is immediately superseded and archived to prevent its accidental use.
This is particularly critical for documents like the Investigator’s Brochure or the informed consent form. Using an outdated version of either is a major compliance deviation with significant ethical implications.
An audit trail is the definitive proof of control. It is a secure, computer-generated, time-stamped log that records who performed what actions, and when, for any given document. During an inspection, this is the primary record reviewed to verify data integrity and procedural compliance. A complete and accurate audit trail is essential for a defensible position.