In clinical trials, change is a constant. Protocol amendments are not a possibility; they are an operational reality. As new safety data emerges or the scientific basis of a study evolves, the protocol must adapt. This adaptation initiates a cascade of updates across study documentation, with the Informed Consent Form (ICF) being a primary consideration.
Managing these ICF updates while a trial is ongoing is a significant operational and regulatory challenge. The process extends beyond simple document editing; it requires a robust, documented system that demonstrates control over the study and, most importantly, protects trial participants.
The Reality of Change in Clinical Trials
For professionals in clinical operations, regulatory affairs, or medical writing, establishing a proficient workflow for ICF updates is a key indicator of operational excellence. It is a complex process involving multiple functional areas and demanding a high degree of precision.
Protocol Amendments Are an Inevitable Part of Study Conduct
Protocol amendments are a standard component of clinical trial management, particularly in long-term, complex studies. This is substantiated by published data. A comprehensive review in the Journal of the American Medical Association found that 66% of trials involved substantive protocol changes that often necessitated modifications to the informed consent language.
Notably, for Phase III trials, over a third of these changes occur after the first participant is enrolled. This context means that clinical trial teams are continually managing critical document updates while concurrently recruiting and monitoring participants. An improvised, ad-hoc approach to this process introduces risks such as version control errors, delayed Institutional Review Board (IRB) submissions, and inconsistent implementation across sites.
A well-defined ICF update process is not merely a compliance activity. It is a direct reflection of a sponsor's commitment to participant safety and data integrity, aligning with the quality management system principles outlined in Good Clinical Practice (GCP) guidelines such as ICH E6(R3).
The Rationale for a Structured Workflow
A structured workflow for managing ICF updates establishes a systematic, logical sequence of events for every change. This approach ensures that each revision is managed with appropriate oversight.
This ensures that every revision is:
- Justified: Each edit in the ICF can be traced directly to a specific protocol amendment.
- Reviewed: All updates undergo formal review and approval by designated personnel before submission to an ethics committee.
- Controlled: A stringent version control system is implemented to prevent the use of outdated forms.
- Communicated: Clinical sites receive clear, unambiguous instructions regarding the implementation of the new ICF.
Ultimately, implementing such a structured process creates a transparent and defensible audit trail, contributing to a state of continuous inspection readiness.
Why Protocol Amendments Drive ICF Changes
The Informed Consent Form (ICF) functions as a direct communication to the trial participant, with its content derived entirely from the clinical trial protocol. It is not a standalone document; its content is based on the protocol, the Investigator's Brochure (IB), and other essential trial documents. Consequently, any modification to the protocol necessitates an evaluation of its impact on the information provided to participants.
This evaluation is the starting point for managing ICF updates mid-trial. The process should begin as soon as a protocol amendment is contemplated. Whether the change involves a minor adjustment to a visit schedule or a significant addition like a new biomarker assessment, its effect on the participant's understanding of the study must be carefully considered.
Identifying Triggers for an ICF Revision
Certain protocol amendments are clear triggers for an ICF update. These changes introduce new information that could reasonably influence a participant's decision to continue their involvement in the trial.
Common triggers include:
- New Safety Information: An update to the Investigator's Brochure revealing a new or more frequent adverse event is a definitive trigger, directly impacting the "Risks" section of the ICF.
- Changes to Study Procedures: The addition of procedures such as an extra blood draw, a new imaging scan, or a biopsy alters the participant's commitment and must be explicitly described.
- Adjustments to Study Schedule: Requiring more site visits or extending the trial's duration directly affects a participant's time and must be consented to.
- Modifications to Financial Considerations: Any change to compensation or reimbursement for trial-related expenses must be communicated transparently in a revised consent form.
This process flow illustrates how a change in trial conduct leads to a protocol amendment, which, in turn, frequently necessitates an ICF update and potential re-consent.
This visual representation underscores that the protocol amendment is the critical link between a trial's evolution and the ethical obligation to keep participants fully informed.
The Requirement for a Defensible Audit Trail
For every modification made to an ICF, there must be a clear, logical justification that traces it back to a specific section of the protocol amendment. This is a fundamental regulatory expectation. Regulatory agency inspectors will scrutinize the connection between protocol versions and their corresponding ICF versions.
A robust audit trail includes maintaining a detailed "summary of changes" document for the ICF.
This log must specify not only what was changed but also why it was changed, referencing the specific protocol amendment and section. For example: "ICF Section 4.2 (Risks): Added 'headache (common)' based on new safety data in IB v3.0, as cited in Protocol Amendment 2, Section 5.1."
This level of detail creates a transparent narrative of the document's evolution, demonstrating a controlled, deliberate process and adherence to GCP obligations to ensure consent is based on current information.
Synchronizing Documentation Workflows
The ICF update workflow should commence concurrently with the drafting of the protocol amendment, not after its finalization. Involving the medical writing or clinical operations team responsible for the ICF in the amendment review cycle from the outset is an operational best practice.
Running these processes in parallel helps prevent delays and inconsistencies. A common pitfall is securing approval for a protocol amendment, only to discover that the ICF submitted to the IRB/IEC does not accurately reflect the approved changes. Such a discrepancy can lead to submission rejection, delays, and significant compliance issues. Mastering the process of managing protocol amendments across clinical documentation is a foundational skill for teams committed to conducting compliant clinical trials.
Navigating Regulatory and Ethics Committee Submissions
Once a revised Informed Consent Form (ICF) is finalized internally, the next step is securing approval from regulatory authorities and all relevant Institutional Review Boards (IRBs) or Independent Ethics Committees (IECs). This is a formal process demonstrating adherence to participant safety and compliance standards.
An incomplete or poorly organized submission package can lead to significant delays, creating operational and compliance challenges across study sites. The objective is to provide reviewers with a clear and comprehensive account of what changed, the rationale for the change, and its link to the protocol amendment. Ambiguity or missing documentation often results in queries and extends the review timeline.
Assembling a Comprehensive Submission Package
An effective submission package anticipates and addresses reviewer questions. While specific requirements may vary by committee, a standard package for an ICF update typically includes the following core documents.
Key components are:
- Summary of Changes: This document is essential. It must clearly and concisely list every change made to the ICF and link each change to the specific part of the protocol amendment that prompted it.
- Track-Changes Version: A "redlined" version of the ICF provides a visual representation of all additions, deletions, and modifications, enabling reviewers to quickly identify changes.
- Clean Version: This is the final version of the updated ICF intended for implementation upon committee approval.
Providing all three documents creates a transparent audit trail and facilitates an efficient review process.
Good Clinical Practice guidelines (ICH E6) mandate that any change to the ICF must receive IRB/IEC approval before it can be used. The submission package serves as the official record of compliance with this fundamental rule.
Addressing Local Submission Requirements
In multi-regional trials, submission processes can vary significantly. A one-size-fits-all approach is often insufficient. An IRB in one country may have different portal requirements and review timelines than an ethics committee in another.
For instance, some jurisdictions may require translation of the entire submission package, while others may accept key documents in English. Submission methods can range from sophisticated online portals to email-based systems. Early engagement with local regulatory teams or CRO partners is crucial to understand and meet these specific requirements.
An Operational Example
Consider a global Phase III trial where an update to the Investigator's Brochure identifies a new cardiovascular risk. The sponsor team prepares a submission for a central IRB but omits the track-changes version of the ICF, sending only the summary and clean copy.
The IRB returns the submission with a query, requesting the redlined document to verify the changes. This oversight results in a two-week delay. During this period, sites must continue using the old ICF. Consequently, new participants are consented without being informed of a newly identified risk, creating a serious compliance and safety issue.
This scenario highlights how a seemingly minor documentation error can have a direct and significant impact on participant safety and study integrity. Successful submissions depend on meticulous preparation and a thorough understanding of the procedural requirements of each regulatory body and ethics committee.
Version Control: The Foundation of Audit Readiness
In managing informed consent updates during an ongoing trial, disciplined version control is a critical practice for maintaining compliance. This extends beyond file naming conventions to a systematic process ensuring that only the single, correct, IRB/IEC-approved version of the Informed Consent Form (ICF) is in use at clinical sites.
Inadequate version control is a common and avoidable audit finding. Filenames such as ICF_v2.0_final_final.docx in a Trial Master File (TMF) indicate an operational risk. Such naming conventions create confusion, lack auditable metadata, and increase the likelihood of using an obsolete document. A robust system moves beyond ambiguity to establish a formal, trackable process for every iteration of the ICF, from initial draft to approved final version.
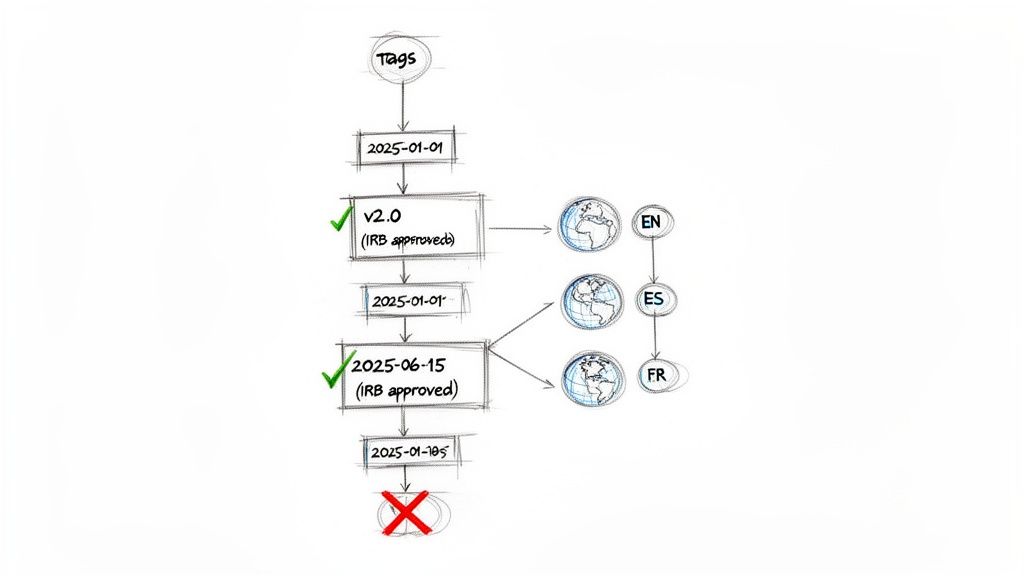
This structured approach is essential for an inspection-ready Trial Master File (TMF/eTMF). It provides clear, undeniable evidence of control over critical study documents.
Components of a Formal Version Control System
An effective version control strategy for ICFs must be logical, consistent, and documented. It should provide a clear and unambiguous history of the document's lifecycle.
Essential components include:
- A Unique Version Number: A clear numbering scheme (e.g., v1.0, v1.1, v2.0) that distinguishes minor administrative updates from major revisions requiring new IRB/IEC approval.
- A Version Date: The date the version was finalized or approved, establishing a clear timeline of changes.
- A Status Indicator: Labels such as "Draft," "In Review," "Approved," or "Superseded" to prevent the use of a document not yet authorized for use.
- A Detailed Change History: A log or table within the document that captures what changed, why it changed, who approved it, and when.
This level of detail elevates version control from a simple filing task to a core component of the quality management system. For a deeper examination of these principles, see our guide on version control best practices for clinical trial documents.
Regulatory Context: Under Good Clinical Practice (ICH E6), sponsors are responsible for implementing and maintaining quality assurance systems. A meticulous ICF version control process is a direct and tangible means of fulfilling this core regulatory obligation, thereby protecting both data integrity and participant safety.
The following table outlines the essential elements an ICF version control system must possess to withstand regulatory scrutiny.
Table: Essential Elements of an ICF Version Control System
| Component | Description | Regulatory Rationale (ICH E6) |
|---|---|---|
| Unique Identifier | A distinct number and/or date for each version (e.g., ICF_PROT123_v3.0_15-Oct-2024). | Ensures unambiguous identification of documents, a key aspect of maintaining a reliable audit trail (Section 8.1). |
| Version History Log | A chronological record of all changes, including the date, author, description of the change, and rationale. | Provides a clear audit trail of document modifications, demonstrating control over the trial's conduct (Section 5.5.3). |
| Document Status | Clear labels like "Draft," "Approved," "Superseded" to define the document's current state in its lifecycle. | Prevents the use of unapproved or outdated documents, protecting participant rights and data integrity (Section 4.8.2). |
| Approval Records | Documented evidence of IRB/IEC approval for each substantive version, linked directly to the version number. | Fulfills the requirement that all trial-related documents receive IRB/IEC approval before implementation (Section 3.1.2). |
| Effective Date | The specific date on which the approved version is authorized for use at clinical sites. | Ensures proper implementation timing and prevents premature use of a new ICF (Section 8.3.2). |
| Superseded Date | The date on which an older version was officially replaced and should no longer be used. | Prevents continued use of outdated forms, which is a critical compliance and safety concern (Section 8.3.2). |
Implementing these components ensures the creation of a defensible, transparent, and compliant system.
The Added Complexity of Translated Versions
In global trials, version control becomes more complex. A common error is failing to maintain a clear, documented link between the master English ICF and its translations. Each translated document requires its own version control but must remain tied to the source document.
The workflow should be as follows: once the master English ICF v2.0 is approved, it becomes the single source of truth. Each translation team must then update their respective documents to precisely mirror the approved changes.
The versioning for a translated document should explicitly state this relationship. For example: "Spanish ICF v2.0 (based on English Master v2.0, approved 15-Oct-2024)." This explicit link is crucial for the TMF, as it demonstrates to an inspector that every language version is consistent with the IRB/IEC-approved master. Any gap in this linkage creates a significant compliance risk.
Version Control and Public Accountability
Meticulous version control is no longer solely an internal best practice. Regulatory authorities are strengthening the connection between internal documentation and public transparency mandates.
For instance, under upcoming 2025 amendments to the FDAAA 801 Final Rule, sponsors will be required to submit a redacted version of the ICF used to enroll participants to ClinicalTrials.gov. This requirement makes it critical to know with certainty which ICF version was used for each participant. Without a disciplined internal version control system, identifying and submitting the correct document becomes a high-stakes, resource-intensive task. Strong version control is the foundation for both regulatory inspection readiness and meeting public accountability obligations.
Implementing the New ICF at Clinical Sites: Communication and Training
An IRB-approved ICF update is not effective until it is correctly implemented at clinical sites. The successful rollout of a new ICF version depends on a robust process for communication, training, and the retirement of previous versions.
This phase is where meticulous version control and regulatory documentation translate into practice. The objective is to create a closed-loop system: distribute the new ICF, obtain confirmation of receipt and understanding from site staff, and verify the archival of the obsolete version. A breakdown in this process poses a risk to participants and data integrity.
Crafting Clear Communications for Clinical Sites
Communication to Principal Investigators (PIs) and Clinical Research Coordinators (CRCs) must be clear, actionable, and unambiguous. Vague instructions can lead to delays or the continued use of an outdated form.
A standard communication package should include:
- A Clear Subject Line: For example, “Action Required: New Approved ICF Version 3.0 for Protocol XYZ-123.”
- The Correct Documents: Attach the final, IRB-stamped clean version of the ICF. Including a summary of changes is also a best practice.
- A Specific "Go-Live" Date: State precisely when the new ICF must be used, which is typically "immediately upon receipt and completion of training."
- Step-by-Step Instructions: Outline the required actions—confirm receipt, train staff, and remove old versions from use.
The goal is to eliminate ambiguity and facilitate prompt and accurate implementation by busy site staff.
Documenting Site Training and Acknowledgment
Distribution of the new ICF is only the first step. Documented proof that the site received the document and that relevant personnel were trained on the changes is essential. This documentation is a cornerstone of the Trial Master File (TMF) and is a primary focus during audits.
A common and avoidable audit finding is the absence of a record of site staff training on a new ICF version before its implementation. A simple training log, signed and dated by the PI and coordinators, closes this compliance gap and demonstrates adherence to Good Clinical Practice (GCP).
The training itself can be scaled to the magnitude of the change. For a minor textual correction, a review of the "summary of changes" document by the CRC and PI, followed by a signed acknowledgment, may suffice. For more significant updates, a brief teleconference may be warranted. The role of this process within the broader context of study management is further explored in our guide on roles and responsibilities in clinical trial documentation.
Retrieving and Archiving Obsolete ICF Versions
Ensuring that all copies of the old ICF are removed from circulation is a critical and often overlooked step. The presence of an old, unstamped ICF in a clinic's active files is a significant compliance issue for any inspector.
Instructions to the site must be prescriptive:
- Immediately locate and collect all physical copies of the old ICF version.
- Transfer these copies to a designated archive for retired study documents.
- Delete any electronic copies from local desktops or shared drives to ensure only the current version is accessible.
This process prevents accidental use of the wrong form and demonstrates control over study documents. While logistically challenging in large, multi-site trials, it is a non-negotiable requirement.
Frequently Asked Questions About ICF Updates
Even with well-defined Standard Operating Procedures (SOPs), questions arise during the ICF update process. A clear understanding of Good Clinical Practice (GCP) and established documentation workflows is key to managing these situations effectively.
Below are some of the most common questions related to this process.
What is the distinction between an administrative and a substantive change?
This distinction is critical as it determines the required submission and review pathway. The primary criterion is whether the change could potentially affect a participant's decision to continue in the trial.
An administrative change is a minor correction or update that does not alter the scientific or ethical aspects of the study. Examples include correcting typographical errors, updating a sponsor's address, or reformatting a page for readability. These changes do not affect a participant's understanding of the risks, benefits, or study requirements.
A substantive change introduces new information that could reasonably influence a participant's continued consent.
The guiding principle, derived from ICH E6, is that new information relevant to a participant's consent must be communicated. This is the definition of a substantive change, which almost always requires a full IRB/IEC review and approval before the revised form can be used.
Examples of substantive changes include:
- Identification of a new risk in an Investigator’s Brochure update.
- Addition of a new mandatory procedure, such as an extra blood draw.
- An extension of the study duration or an increase in the number of required site visits.
While some IRBs may permit notification for administrative changes, substantive changes necessitate a formal submission package, including a summary of changes, redlined versions, and clean copies for review.
How are translated ICFs managed when the English "master" version changes?
This is a significant compliance consideration. When a master English ICF is updated and approved, it initiates a critical workflow for all translated versions. The approved English document serves as the single source of truth.
The process must be tightly controlled. Upon approval of the new English master by the IRB/IEC, qualified medical translators must update each translated version to match it precisely. Quality control steps, such as back-translation or review by a second linguist, are best practices to ensure accuracy.
Each translated ICF requires its own version control that clearly links back to the master. A file name such as "Spanish ICF v2.0 (from English Master v2.0 - 15 Oct 2024)" provides this necessary traceability for the audit trail. Each updated translation must then be submitted to its local ethics committee for approval before it can be implemented at a clinical site.
What documentation does an auditor look for in the TMF for an ICF update?
An auditor reviewing the Trial Master File (TMF/eTMF) should be able to reconstruct the entire history of an ICF update without needing to ask questions. A well-maintained TMF tells a complete, chronological story for each version.
An auditor-ready package for an ICF update must include:
- The final, approved protocol amendment that initiated the update.
- A complete version history for the ICF, detailing all revisions.
- All correspondence with the IRB/IEC, including the submission, summary of changes, official approval letter, and the IRB/IEC-stamped copy of the new ICF.
- Documentation of distribution of the new version to all clinical sites.
- Records confirming that site staff were trained on the new form before it was implemented.
- Evidence that all obsolete ICF versions were either collected from sites or that their destruction or archival was confirmed.
The presence of all these components demonstrates control over the process and adherence to GCP.
How can a centralized documentation platform support this process?
Managing these workflows using spreadsheets and email can introduce significant risk. A centralized documentation platform, such as Skaldi, is designed to serve as the single source of truth for all study documents, including ICFs.
Such a platform can connect related documents, such as the protocol, Investigator's Brochure, and ICFs. When a change occurs in one document, it can automatically flag others that require updates. It enforces version control, preventing sites from accidentally accessing and using an outdated form.
Furthermore, a platform can manage the entire review and approval workflow, automatically generate a comprehensive audit trail, and centralize all related communication. This approach reduces the potential for human error associated with manual processes and improves inspection readiness by ensuring process consistency and documentation integrity.