Maintaining the Trial Master File (TMF) in a state of completeness and inspection readiness is a continuous operational discipline throughout the clinical trial lifecycle. The objective is to manage the TMF actively so that it is complete, timely, and of high quality at any given moment. This proactive approach ensures the study's conduct can be reconstructed for regulatory authorities upon request.
The Three Pillars of TMF Health
A well-maintained TMF should provide a clear and accurate account of the study's conduct. Its integrity rests on three foundational pillars: completeness, timeliness, and quality. These are not merely operational best practices but fundamental standards for regulatory compliance and inspection readiness.

Regulatory bodies view the TMF as the definitive record demonstrating that a trial adhered to Good Clinical Practice (GCP). Guidelines such as ICH E6(R3) specify that the TMF must contain all essential documents. This requirement extends beyond simple storage; it is intended to enable a comprehensive evaluation of the trial's conduct and the integrity of the data generated.
The TMF is a dynamic file that must contemporaneously mirror the trial's progress. When regulatory agencies like the FDA or EMA conduct an inspection, they expect a coherent and complete narrative, not a collection of disconnected documents.
Consequences of an Unhealthy TMF
A TMF with significant gaps, chronically late filings, or poor-quality documents is a major concern during an inspection. Such issues are frequently cited as critical findings because they obscure the trial narrative, raise doubts about data integrity, and suggest a lack of sponsor oversight.
For sponsors and CROs, the consequences can be significant. A deficient TMF may delay a regulatory submission, necessitate resource-intensive remediation projects, or, in a worst-case scenario, lead to the rejection of study data. Therefore, maintaining TMF health is a critical component of an organization's overall risk management strategy.
Inspection readiness is not achieved by last-minute efforts before an audit. It is a state maintained through consistent and disciplined TMF management throughout the study. Documents must be filed contemporaneously as they are created or finalized.
The following table summarizes the core components of TMF health.
The Three Pillars of TMF Inspection Readiness
| Pillar | Definition | Regulatory Driver (Example) | Operational Impact |
|---|---|---|---|
| Completeness | All expected essential documents, as defined by the study-specific TMF Index, are present in the TMF. | ICH E6(R3) 5.5.3: The sponsor should ensure that the TMF is complete. | A complete TMF allows for the full reconstruction of the trial, demonstrating proper conduct and data integrity. Missing documents create critical gaps in the study narrative. |
| Timeliness | Documents are filed in the TMF promptly after they are generated, finalized, or received. | MHRA Guidance: Expects documents to be filed in a "timely manner" to reflect the trial's progress contemporaneously. | Prevents large backlogs before milestones and ensures the TMF accurately reflects the trial’s current state at any point, a key expectation for inspectors. |
| Quality | Every document is legible, accurate, correctly versioned, and properly executed (e.g., signed, dated, with complete metadata). | 21 CFR Part 11: Requires that electronic records be accurate and reliable. | Poor quality (e.g., illegible scans, missing signatures) can render a document invalid, undermining its evidentiary value even if it is filed on time. |
Internalizing these principles helps foster a culture where TMF health is a shared responsibility across the entire study team.
Achieving a State of Continuous Readiness
Embedding TMF management into core clinical operations is essential for achieving a continuous state of readiness. This begins with a comprehensive understanding of how these three pillars interrelate.
Practical considerations include:
- Completeness: The definition of "complete" must be established for each specific study via a TMF index. A common framework for building a comprehensive and standardized index is the DIA Trial Master File Reference Model.
- Timeliness: This requires operational discipline. Documents should be filed shortly after they are finalized to keep the TMF current and avoid pre-inspection resource strain.
- Quality: TMF quality must be maintained. A TMF can be quantitatively complete yet fail an inspection due to poor-quality records. Each document must be legible, accurate, and properly executed with required signatures and dates.
However, achieving confidence in these areas presents an industry-wide challenge. A 2021 survey indicated that only 6% of sponsors and CROs felt their TMF metrics were 'very accurate,' while 53% rated them as 'somewhat accurate.' This highlights a significant gap in visibility. Effective management requires accurate measurement, and without a clear, real-time view of TMF status, continuous inspection readiness remains an elusive goal.
Building Your Framework for Continuous Readiness
Sustained TMF completeness and inspection readiness are the results of a well-defined operational framework implemented from study start-up. A reactive approach, characterized by scrambling before an inspection, often leads to critical findings. The objective is to integrate TMF management into the daily fabric of clinical operations, making it a continuous and proactive process.
This process begins with a comprehensive TMF management plan, which serves as the operational playbook for the study. It must detail not just what is filed, but how, when, and by whom. This plan should be a living document, tailored to the specific protocol and operational model, whether the trial is managed in-house, outsourced to a CRO, or uses a hybrid approach.
An effective plan connects the creation of key documents, such as the protocol or informed consent forms, directly to their filing requirements. This perspective ensures documentation is treated as an integrated and critical step in the trial lifecycle, rather than a separate administrative task.
Defining Document Ownership and Accountability
Ambiguous responsibility is a common failure point in TMF management. The TMF plan must mitigate this risk by assigning clear, explicit ownership for every document type or artifact. This involves mapping roles to specific stages of the document lifecycle.
For example, the plan must specify which role is responsible for obtaining and filing final, signed investigator agreements from all sites—whether it is the CRA, a central study start-up lead, or a TMF specialist. This level of detail leaves no room for misinterpretation.
An inspector needs to see a clear chain of custody for every essential document. The TMF plan, along with tools like delegation logs, should precisely define who was responsible for creating, reviewing, approving, and ultimately filing each component of the trial's story.
This accountability framework must extend to all partners involved in the trial. A well-designed plan clearly delineates responsibilities between the sponsor, the CRO, and any third-party vendors.
- Sponsor: Typically retains ownership of high-level documents like regulatory correspondence and the final clinical study report.
- CRO: Often manages day-to-day site management documents, such as monitoring visit reports and site communication logs.
- Central Lab: Is responsible for providing its own essential documents, including lab certifications and normal range documentation.
Without this clarity, documents can be overlooked, leading to significant gaps discovered during last-minute reviews.
Establishing the TMF Index and Filing Timelines
The TMF index, often based on the DIA TMF Reference Model, serves as the detailed table of contents for the study. It must be customized to reflect the trial's unique requirements, including any country-specific or study-specific documents. The index defines what "complete" means for the TMF and serves as the master checklist for completeness reviews.
Once the index is established, the plan must define realistic and firm filing timelines. Regulatory bodies like the MHRA expect documents to be filed in a "timely manner" to contemporaneously reflect the trial's progress. Significant delays in uploading documents are a red flag for inspectors, who can easily review upload dates in an eTMF system. The selection of an electronic trial master file software should support compliant and timely workflows.
An example of timeliness targets includes:
- Monitoring Visit Reports: To be filed within 10 business days of report finalization.
- Site Staff Training Records: To be filed within 5 business days of the training session.
- Signed Informed Consent Forms (example first page): To be filed within 3 business days of the subject's visit.
These deadlines create a rhythm of compliance that helps prevent the accumulation of a large TMF backlog. This proactive approach supports a state of continuous readiness, demonstrating to regulatory authorities that the trial is under control and properly overseen.
Using Metrics to Monitor TMF Completeness
Achieving inspection readiness requires more than subjective assessments. Moving from a reactive approach to a state of continuous readiness depends on measuring relevant metrics. Key Performance Indicators (KPIs) provide a data-driven, real-time view of TMF health.
Effective monitoring involves using insightful metrics to assess the true state of trial master file completeness and inspection readiness. These KPIs can function as an early warning system, identifying process issues before they escalate into critical inspection findings.
Moving Beyond Basic Document Counts
A high-level completeness percentage provides an initial snapshot but does not tell the whole story. A TMF could be 95% complete, but if the missing 5% includes the signed protocol or initial IRB approvals, significant compliance issues exist. Tracking more granular, process-focused metrics is essential.
Key metrics for TMF managers include:
- Document Cycle Times: The time elapsed from document finalization to filing in the TMF. Prolonged cycle times can indicate process bottlenecks or insufficient prioritization of TMF filing.
- Quality Control (QC) Pass/Fail Rates: The percentage of documents rejected during the QC check. High rejection rates may signal training deficiencies or inconsistent quality standards.
- Timeliness Adherence: The percentage of documents filed within the timelines defined in the TMF plan. This data can be analyzed by document type, site, or functional area to pinpoint problem areas.
A Practical Scenario in a Multi-Center Trial
Consider a trial with 50 active sites. By tracking timeliness KPIs for Site Initiation Visit (SIV) documentation, a trend emerges: while 45 sites filed their SIV packages within the required 10 business days, five sites are significantly delayed, with some exceeding 30 days.
A simple overall completeness metric would have obscured this issue. However, this specific KPI indicates a problem that requires further investigation. It prompts questions about the workload of the CRAs for those sites or potential misunderstandings of the process. This targeted data allows for intervention before an inspector discovers the issue.
Inspectors are increasingly data-savvy and may analyze eTMF metadata for trends. A high volume of document uploads immediately preceding an inspection suggests that the TMF was not managed contemporaneously.
Setting Realistic Tolerance Limits for Completeness
While 100% perfection across thousands of documents is not always feasible, regulators expect a risk-based approach to quality. Establishing realistic tolerance limits in the TMF management plan is therefore critical. These limits define acceptable deviations from completeness, timeliness, or quality targets.
The tolerance level should be commensurate with the criticality of the document. For example, based on historical regulatory expectations reflected in EMA inspection reports, the tolerance for a missing "stand-alone" essential document like the clinical trial protocol is typically 0%. For high-volume documents, such as investigational product shipment records across numerous sites, a tolerance of <1% might be considered reasonable.
This risk-based distinction helps the team focus its oversight on documents that are most critical to trial integrity and patient safety.
The following table provides examples of core metrics and potential targets that can be adapted for a TMF plan.
Essential TMF Health Metrics and KPIs
Tracking relevant KPIs is fundamental to assessing TMF health. These metrics provide objective evidence of contemporaneous management and proactive oversight, both of which are key areas of focus for inspectors.
| Metric Category | KPI Example | Target/Tolerance | Rationale for Inspections |
|---|---|---|---|
| Completeness | % of expected essential documents filed per TMF index | Overall: >95% Critical Docs: 100% |
Demonstrates the TMF can fully reconstruct the trial. Gaps in critical documents are a significant finding. |
| Timeliness | % of documents filed within 15 days of finalization date | >90% | Shows the TMF is managed contemporaneously and reflects the trial's real-time progress, not just historical activity. |
| Quality | First-pass QC acceptance rate | >98% | A high rate indicates robust upstream processes and well-trained staff, reducing rework and ensuring document reliability. |
| Oversight | Frequency of documented sponsor TMF reviews | Quarterly (minimum) | Provides tangible evidence of sponsor oversight, a key requirement under ICH GCP, especially in outsourced models. |
By consistently tracking these metrics, TMF management evolves from a reactive, administrative exercise into a proactive quality oversight function. This not only builds a more robust TMF but also fosters a culture of inspection readiness across the study team.
A Practical Guide to Remediating TMF Gaps
Discovering a gap in the Trial Master File is an operational reality. What matters to inspectors is not the existence of the gap itself, but the robustness and transparency of the process used to address it.
A well-documented remediation effort can demonstrate a functional quality management system and procedural control. The objective is to move from reactive problem-solving to a structured, compliant resolution process. This process should begin as soon as a deficiency—such as a missing document, incorrect version, or incomplete record—is identified. Simply uploading the correct file without context is insufficient; the entire correction must be documented.
Implementing a Formal Remediation Process
An effective gap resolution is based on a formal, documented process, often managed through a Corrective and Preventive Action (CAPA) plan. A CAPA provides a structured framework that is both auditable and defensible, transforming an error into an opportunity to demonstrate a commitment to quality.
A systematic approach includes several key stages:
- Identification and Logging: The gap is formally logged with specific details about what is missing or incorrect, its expected location, and how it was discovered.
- Root Cause Analysis (RCA): This critical step investigates why the gap occurred. The cause could be a system failure, an ambiguous process, or a training issue.
- Corrective Action: This is the immediate fix, which might involve locating the correct document, obtaining necessary signatures, or correcting metadata in the eTMF.
- Preventive Action: This is the long-term solution designed to strengthen the system, such as updating an SOP, retraining staff, or reconfiguring an eTMF workflow to prevent recurrence.
- Documentation: Every step, from the RCA to final verification, must be meticulously documented in a note-to-file or within the CAPA system.
The dashboard below illustrates the health metrics that a robust remediation process aims to restore.
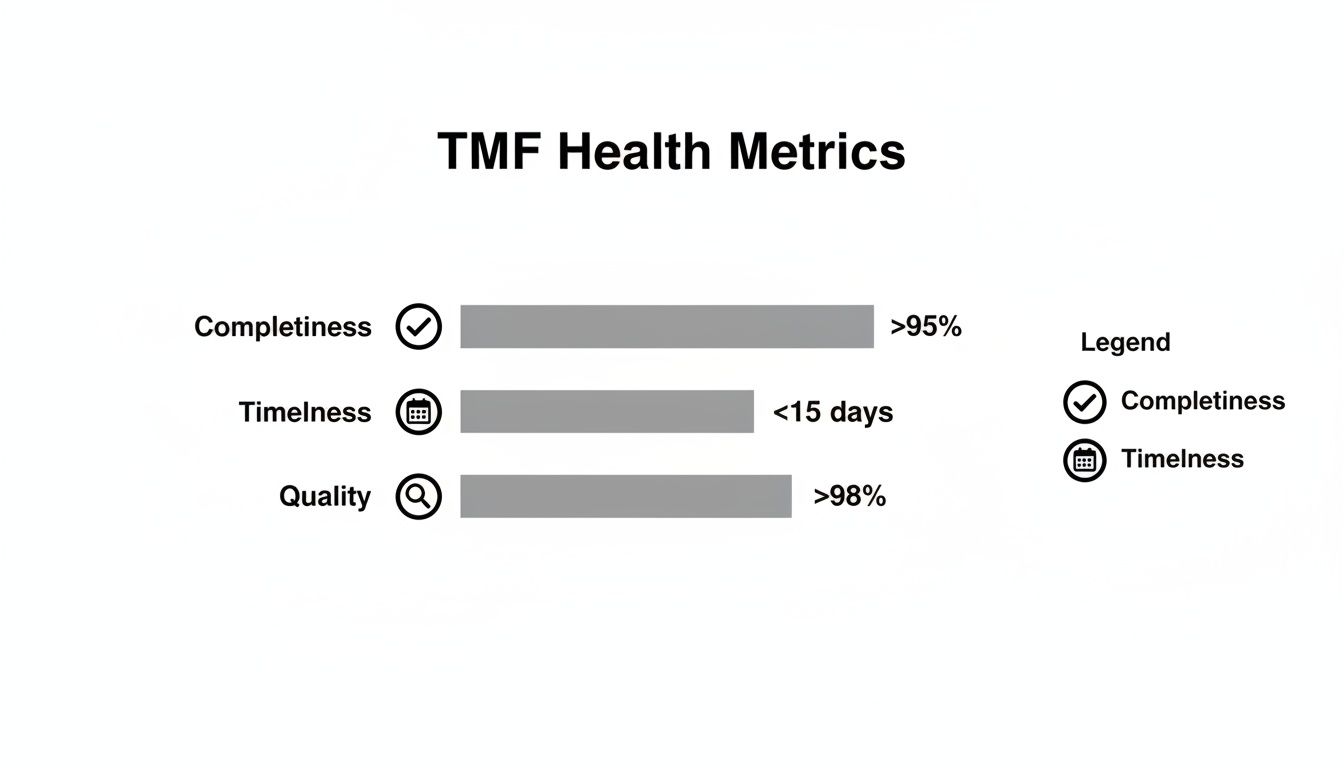
Meeting targets for completeness, timeliness, and quality is the primary goal. A structured remediation plan is the mechanism for getting back on track when deviations occur.
Real-World Remediation Scenarios
The following examples illustrate how this process applies to common TMF issues. The context and rationale behind the remediation are as important as the fix itself.
Scenario 1: Missing Delegation of Authority (DoA) Log
A routine TMF review reveals a missing DoA log for a site. This is a critical document linking study tasks to qualified personnel.
- Root Cause Analysis: The investigation reveals that a new site coordinator mistakenly believed the log was only required after the first subject was enrolled, indicating a gap in the site initiation training process.
- Corrective Action: The CRA contacts the site, clarifies the requirement, and works with the Principal Investigator to complete and sign the DoA log. The log is dated to reflect the staff's actual involvement dates to ensure accuracy.
- Preventive Action: The sponsor updates its Site Initiation Visit (SIV) checklist to include a specific verification step for the DoA log and amends training materials for new site coordinators to explicitly cover this requirement.
- Documentation: A detailed note-to-file is created, explaining the finding, the root cause, the corrective actions taken (including the rationale for the dates used), and the preventive measures implemented.
Scenario 2: Investigator’s Brochure (IB) Version Control Issue
A quality check identifies that two sites have acknowledged receipt of IB Version 4.0, but there is no record of them receiving Version 3.0.
- Root Cause Analysis: The clinical trial assistant responsible for IB distribution overlooked these two sites during the Version 3.0 mailing due to a manual tracking error in a spreadsheet.
- Corrective Action: The sites are contacted to confirm they have and are using the current Version 4.0. They are also sent Version 3.0 with a request to acknowledge its receipt retrospectively to complete the TMF record.
- Preventive Action: The distribution process is revised to use an automated tracking system that requires positive confirmation from each site before a distribution is marked as complete.
- Documentation: A memo is filed in the TMF detailing the oversight, the sites involved, confirmation that the current IB is in use, and the procedural update designed to prevent future errors.
The narrative built around a remediation is crucial. An inspector is more likely to accept a well-documented mistake coupled with a thoughtful correction and process improvement than an unexplained gap or a hastily uploaded document with no context.
Regulatory scrutiny of TMFs has increased, with agencies frequently citing failures in completeness and timeliness. Analyses indicate that timeliness lapses occur in 15-25% of multi-site trials, and TMF transfers from a CRO to a sponsor can result in completeness dropping by 10-20%. These figures underscore the importance of robust oversight and a defined remediation process.
More information on these regulatory trends and their impact on trial master file completeness and inspection readiness can be found in PharmaLex's analysis of TMF quality. A consistently applied, structured remediation framework builds a resilient TMF that can withstand inspection.
What to Do When the TMF Inspector Arrives
The notification of an inspection from a regulatory agency is the culmination of TMF readiness efforts. When an inspector arrives, the goal is to confidently present the complete story of the trial as documented in a well-maintained TMF.

A successful inspection depends on a prepared team executing a well-defined plan. The objective is to make the inspector's review as efficient as possible, demonstrating control over documentation and processes. A calm, methodical approach from the outset helps build trust.
Assemble the Inspection Team
First, assemble the core inspection team. Each member must have a clearly defined role to avoid confusion and delays under pressure.
A typical team includes:
- Inspection Lead/Host: The single point of contact for the inspector. This person manages the inspection room, coordinates all document requests, and keeps the process on schedule.
- Subject Matter Experts (SMEs): Representatives from clinical operations, data management, regulatory affairs, and quality assurance who are available to explain specific documents or processes.
- Scribe/Document Runner: The official record-keeper of the inspection. This person logs every request, tracks all documents provided, and takes notes on interactions. This log is an essential record.
- Back Room Support: A team working in a separate area to retrieve requested documents and prepare responses, supporting the front room without directly interacting with the inspector.
Managing the Inspection in Real-Time
The command center, whether physical or virtual, should be prepared with reliable access to the eTMF, essential documents like the protocol, and communication tools. A well-prepared inspection room signals professionalism and readiness.
Once the inspection begins, real-time management is critical. The scribe must log each request as it is made, detailing what was asked for, who is retrieving it, when it was delivered, and any inspector feedback. This log is key to managing follow-ups and tracking the inspector’s areas of focus.
An inspector’s request is a line of inquiry used to understand the trial's narrative. The timeliness and accuracy of the response, including its context, reflect the integrity of the TMF and the organization's command of the study.
This process relies on robust, validated technology. A thorough understanding of data integrity is essential; our guide on FDA 21 CFR Part 11 compliance provides further information on managing electronic records.
Answering Inspector Questions
How questions are answered is as critical as the documents provided. Responses should be factual, clear, concise, and directly address the question. Avoid speculating or volunteering unsolicited information, which can open new lines of questioning.
Best practices for responding include:
- Listen Carefully: Ensure you fully understand the question before answering.
- Answer the Question Asked: Provide a factual answer based on the TMF. If the information is not immediately available, state, "I do not have that information at this moment, but I will get it for you."
- Provide Brief Context: When providing a document, frame it succinctly. For example, "Here is the IRB approval for protocol amendment three, which was implemented to add a secondary endpoint."
- Maintain Professionalism: Remain composed, even when facing difficult questions or a potential finding. A cooperative and professional attitude is important.
Conduct a Mock Inspection
There is no substitute for a rehearsal. Conducting a mock inspection weeks before the actual event is a valuable preparatory exercise. This dry run helps identify process gaps, test the team’s response times, and build confidence.
Engage an independent party to act as the inspector, making requests for a variety of documents. This will test the team's ability to:
- Locate documents in the eTMF quickly under pressure.
- Clearly explain the trial's narrative as they proceed.
- Maintain an accurate and detailed request log.
This practice allows for the identification and correction of weaknesses when the stakes are low. When the real inspectors arrive, the team will be better prepared. Successfully navigating an inspection is the ultimate demonstration of a commitment to trial master file completeness and inspection readiness.
Common TMF Management Questions
Even with a robust plan, day-to-day TMF management can present specific questions. Clarifying these practical issues is key to maintaining a complete and inspection-ready TMF.
What Is the Difference Between TMF Completeness and Quality?
Completeness refers to the presence of documents. It is a quantitative check against the TMF index to confirm whether every required document has been filed.
Quality refers to the content and integrity of those documents. This includes legibility, correct versioning, and the presence of all required signatures and dates. A TMF can be 100% complete quantitatively but fail an inspection if the quality of the documents is poor. Inspectors expect both, as poor quality undermines the integrity of the TMF.
How Often Should We Conduct TMF Completeness Reviews?
The optimal frequency depends on the study's phase, complexity, and risk profile. Formal, in-depth reviews should be conducted at major study milestones, such as First Patient In, after an interim analysis, and before Database Lock.
However, periodic reviews alone are insufficient. Routine reviews, whether monthly or quarterly, are necessary for ongoing TMF health. The industry trend, encouraged by regulators, is toward continuous, real-time oversight. A modern eTMF system can facilitate this by integrating inspection readiness into daily workflows rather than relying on periodic spot-checks.
Who Is Ultimately Responsible for the TMF in a Sponsor-CRO Partnership?
Regulatory guidelines are unequivocal: the sponsor always retains ultimate responsibility for the TMF, even when study and TMF management activities are outsourced to a Contract Research Organization (CRO). This is a fundamental aspect of sponsor oversight under ICH GCP.
While tasks may be delegated, ultimate responsibility cannot. The sponsor must demonstrate active oversight of the CRO’s TMF activities through documentation, such as:
- A detailed TMF plan that clearly defines roles and responsibilities.
- Records of periodic sponsor reviews of the CRO's TMF.
- Formal audits of the CRO's systems and processes.
An inspector will hold the sponsor accountable for any TMF deficiencies identified in a CRO-managed study. This regulatory responsibility cannot be fully transferred, making robust, documented oversight non-negotiable.
What Are Some of the Most Commonly Missed TMF Documents?
While specific to each study, certain types of documents are commonly found to be missing. Awareness of these high-risk areas can help teams focus their oversight.
Commonly missed documents can be categorized by their origin. At the site level, these often include CVs and medical licenses for staff who joined mid-study, outdated Delegation of Authority logs, and temperature excursion reports. Critical but informal site communications are also frequently overlooked.
Centrally-managed documents that are often missing include final, fully-executed vendor contracts, minutes from meetings where key decisions were made, and documentation of training on protocol amendments. The most effective defense against these gaps is a detailed, study-specific TMF index combined with a disciplined, routine QC process that targets these known problem areas.