Regulatory Affairs (RA) serves as the critical interface between pharmaceutical companies and the global health authorities responsible for safeguarding public health. This function is not merely administrative; it is a strategic discipline that guides a therapeutic product through the complex regulatory framework, from initial nonclinical research through to post-market surveillance.
The Strategic Role of Regulatory Affairs in Drug Development
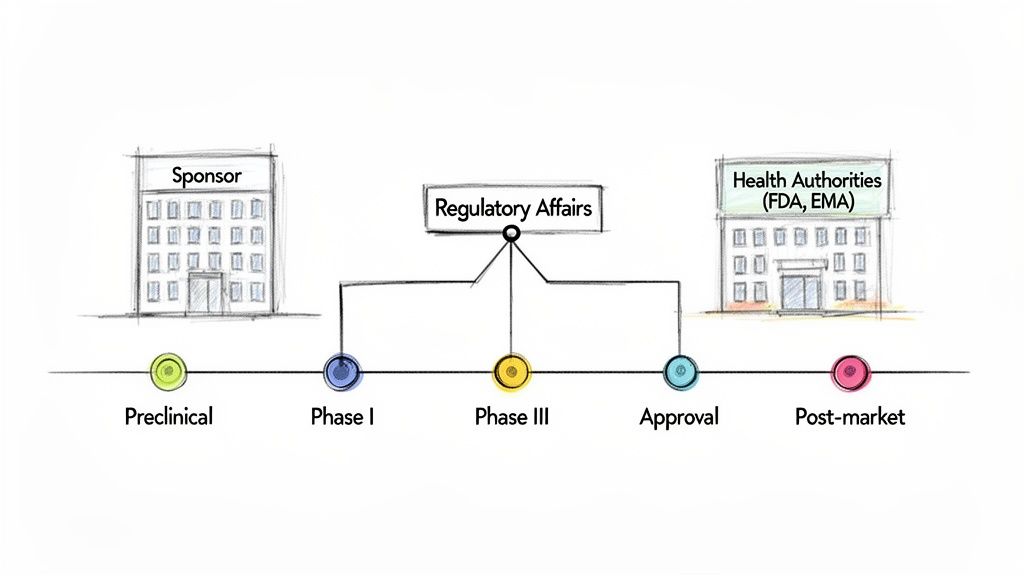
Within the lifecycle of drug development, the Regulatory Affairs department functions as a central coordinating body. Its engagement begins at the earliest stages of product development, ensuring that scientific and clinical strategies align with regulatory requirements from inception. The primary responsibility of RA is to manage all communications and submissions to regulatory bodies such as the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA).
This strategic involvement is critical during the preclinical phase, where RA professionals assess the proposed drug candidate and define the most viable regulatory pathway for approval. They interpret complex regulations and scientific guidelines, translating them into an actionable development plan for research and clinical teams. This early strategic planning influences key decisions, including the design of initial safety studies and the structure of the first-in-human clinical trials.
Guiding Clinical Development and Documentation
As a drug candidate advances to human clinical trials, the strategic role of the RA team intensifies. They are responsible for preparing and submitting the Investigational New Drug (IND) application or Clinical Trial Application (CTA), which is the regulatory prerequisite for initiating human studies. Throughout the trial's duration, RA ensures that all activities comply with Good Clinical Practice (GCP) standards.
The RA function provides critical input and oversight on key clinical trial documents that form the basis of the regulatory submission:
- Clinical Trial Protocols: RA ensures the study design is scientifically robust and meets regulatory expectations for demonstrating safety and efficacy.
- Investigator’s Brochures (IBs): RA verifies that all current nonclinical and clinical data, particularly safety information, is accurately conveyed to investigators.
- Informed Consent Forms (ICFs): RA confirms that the language used is clear, ethically sound, and adheres to all legal and regulatory requirements for patient protection.
- Clinical Study Reports (CSRs): Following trial completion, RA contributes to the structure and content of the final report, ensuring the presentation of results aligns with regulatory standards such as ICH E3.
The core function of Regulatory Affairs is to integrate scientific data with legal and regulatory requirements. The objective is to construct a logical, evidence-based case for the drug that is both scientifically valid and fully compliant, thereby protecting patient safety and the sponsor's investment.
Navigating Toward Market Approval
The ultimate objective for a regulatory affairs team is to secure and maintain marketing approval for a new product. This involves compiling years of accumulated data into a comprehensive submission, such as a New Drug Application (NDA) in the U.S. or a Marketing Authorisation Application (MAA) in Europe. This submission is a meticulously structured argument demonstrating that the drug's benefits outweigh its known risks.
Following submission, the RA team leads all interactions with the health authority, addressing inquiries, clarifying data, and negotiating the final product labeling. The role extends beyond initial approval to manage post-market commitments, pharmacovigilance reporting, and submissions for any product changes, such as new indications or manufacturing updates. In this capacity, RA professionals serve as the lifelong stewards of the product's regulatory compliance.
Navigating the Global Pharmaceutical Regulatory Landscape
Bringing a new drug to the global market requires navigating a complex web of national and regional regulations. Each major market operates under a distinct regulatory authority with its own specific requirements for approving new medicines. Effective regulatory affairs pharmaceutical work necessitates a thorough understanding of this diverse global landscape.
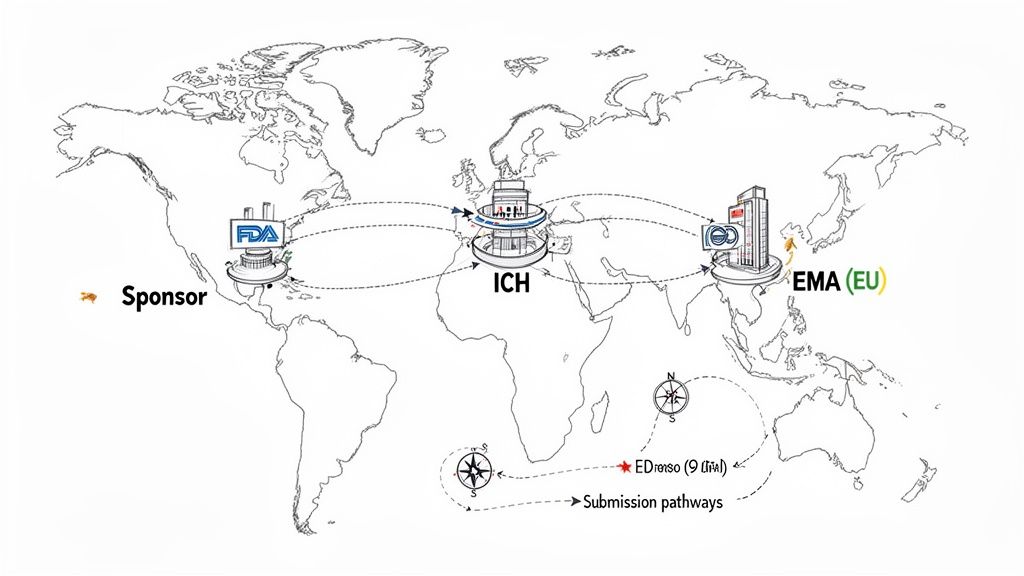
A foundational element of global drug development is the International Council for Harmonisation (ICH). The ICH is not a regulatory body; it is a global initiative that brings together regulatory authorities and the pharmaceutical industry to discuss scientific and technical aspects of drug registration. Its purpose is to achieve greater harmonization in the interpretation and application of technical guidelines and requirements for product registration, reducing the need for duplicative testing.
The Bedrock: ICH Good Clinical Practice
The ICH E6 guideline on Good Clinical Practice (GCP) is a cornerstone of international ethical and scientific quality standards for designing, conducting, recording, and reporting trials that involve human subjects. Adherence to GCP provides public assurance that the rights, safety, and well-being of trial subjects are protected and that the clinical trial data are credible.
For sponsors and Contract Research Organizations (CROs), compliance with ICH E6 is a fundamental requirement. It serves as a universal standard that facilitates the mutual acceptance of clinical data by regulatory authorities in different jurisdictions. We cover this in much more detail in our guide on ICH guidelines for clinical trials.
Meet the Gatekeepers: FDA and EMA
While ICH provides harmonized guidelines, the authority to approve a drug for marketing rests with national or regional health authorities. The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are two of the most influential regulatory bodies globally.
Their distinct roles and mandates are outlined below.
Key Regulatory Bodies and Their Core Mandates
| Regulatory Authority | Region/Jurisdiction | Primary Role in Drug Development |
|---|---|---|
| ICH | Global | Develops harmonized technical guidelines (like GCP) to promote a unified standard for clinical trials. |
| FDA | United States | Directly reviews and approves drugs for the U.S. market through submissions like INDs and NDAs. |
| EMA | European Union | Evaluates drugs for a centralized marketing authorization valid across all 27 EU member states plus Iceland, Liechtenstein, and Norway. |
Each authority has its own submission procedures and regulatory structures. For example, the EMA's centralized procedure allows for a single application to grant marketing authorization across the entire EU, a different operational model compared to the FDA's national approval process.
The growing complexity of these global frameworks has driven significant growth in the field. The global pharmaceutical regulatory affairs market was valued at USD 9.78 billion in 2024 and is projected to reach USD 20.56 billion by 2033. This expansion reflects the increasing demand for professionals skilled in navigating these intricate systems.
A common misconception is that harmonization implies uniformity. While ICH guidelines establish a common foundation, regional authorities maintain unique requirements and preferences for submission content and format.
An effective regulatory strategy anticipates these differences from the outset. A simultaneous submission to both the FDA and EMA requires a core data package robust enough to meet the standards of both agencies, while also accounting for region-specific requirements, such as the Pediatric Investigation Plan (PIP) required by the EMA. Such foresight is essential for a streamlined review process and timely approval.
Mastering Core Clinical Trial Documentation
In pharmaceutical regulatory affairs, documentation serves to construct a cohesive narrative of a product's development, safety, and efficacy. Each core document, from the initial protocol to the final clinical study report, must be logically interconnected to support the overall submission.
These documents are not static; they are subject to a continuous lifecycle of development and revision. A protocol may be amended based on emerging data, while the Investigator’s Brochure is updated annually to reflect new safety information. This disciplined process of drafting, reviewing, and versioning is essential for ensuring that all submissions are prepared to withstand regulatory scrutiny.
The Clinical Trial Protocol: The Study Blueprint
The Clinical Trial Protocol is the operational manual that ensures a study is conducted uniformly across all participating sites. It is developed in accordance with ICH E6 (Good Clinical Practice) to protect the rights of participants and ensure the integrity of the data collected.
Key elements include:
- Study Objectives and Endpoints: Defines the primary and secondary questions the trial aims to answer and the specific measures used to assess outcomes.
- Study Design and Methodology: Details the trial type (e.g., randomized, double-blind), treatment arms, patient enrollment procedures, and data collection methods.
- Safety Monitoring Plan: Outlines the procedures for identifying, evaluating, and reporting adverse events to ensure participant safety.
All subsequent documentation, including the Informed Consent Form and the Clinical Study Report, must be consistent with the protocol. Any deviation or amendment must be formally documented and justified, as the protocol represents a binding agreement with health authorities.
The Investigator’s Brochure: A Comprehensive Profile
The Investigator’s Brochure (IB) is a comprehensive compilation of the nonclinical and clinical data on the investigational product. Its purpose is to provide investigators with the information they need to understand the rationale for the trial and manage patient care appropriately.
- The IB structure adheres to guidelines set forth in ICH E6.
- It is updated at least annually or whenever significant new safety information becomes available.
This living document ensures that investigators and ethics committees have access to the most current information regarding potential risks associated with the product.
The Informed Consent Form: Upholding Ethical Standards
The Informed Consent Form (ICF) operationalizes the ethical principle of informed consent. It translates complex scientific and medical information from the protocol into language that is understandable to potential trial participants. The ICF must clearly explain:
- The study’s purpose, duration, and procedures
- Potential risks and anticipated benefits
- The participant's right to withdraw from the study at any time without penalty
Regulatory authorities and Institutional Review Boards (IRBs)/Ethics Committees (ECs) meticulously review the ICF to ensure it meets global ethical standards (such as ICH GCP) as well as local legal and cultural requirements. For a deeper dive into these essential documents, check out this detailed guide.
The Clinical Study Report: The Final Narrative
Upon completion of a clinical trial, the Clinical Study Report (CSR) synthesizes all study data and analyses. Prepared in accordance with the ICH E3 guideline, the CSR presents the trial's conduct and results in a transparent and structured format.
The CSR is the definitive account of the clinical trial, transforming raw data into a structured narrative that presents a clear argument for the product's safety and efficacy.
A comparison of these key documents highlights their distinct roles:
| Document | Guideline | Main Focus |
|---|---|---|
| Protocol | ICH E6 | Consistent execution |
| Investigator’s Brochure | ICH E6 | Up-to-date risk profile |
| Clinical Study Report | ICH E3 | Final results & analysis |
This detailed reporting is vital in a market projected to reach USD 36.12 billion by 2034, with clinical studies accounting for 48.87% of that value. In oncology alone—holding a 32.07% revenue share—robust documentation like Statistical Analysis Plans and CSRs drives sponsor and CRO strategies. Learn more about this growing market on Precedence Research.
Ultimately, all clinical trial documentation must contribute to a single, consistent, and defensible narrative that meets the rigorous standards of regulatory review.
Building Workflows for Audit Readiness and Compliance
Knowledge of clinical trial documentation requirements is foundational, but operational execution is paramount. In regulatory affairs pharmaceutical operations, audit readiness is not a preparatory phase but a continuous state achieved through robust, predictable workflows. These processes provide the framework for a compliant, inspection-ready operation.
This begins with the standardization of key documents, such as protocols and investigator's brochures, through the use of controlled templates. Templates are a quality control mechanism, ensuring that all necessary regulatory and operational elements are consistently included from the outset. This reduces variability and mitigates the risk of omissions.
From there, clear, auditable review and approval cycles must be established. The roles and responsibilities of all stakeholders—including medical writers, clinical scientists, Quality Assurance (QA), and regulatory affairs—must be explicitly defined. This structured approach prevents operational bottlenecks and ensures that all feedback is systematically captured, addressed, and documented, providing a clear rationale for any changes made.
The Dynamics of Sponsor and CRO Collaboration
Ineffective collaboration between sponsors and Contract Research Organizations (CROs) is a common source of inspection findings. Well-defined processes are therefore a critical risk mitigation tool in these partnerships.
A successful sponsor-CRO relationship is built on several key workflow components:
- Shared Document Repositories: All parties must operate from a single source of truth. A common document management system prevents version control issues, such as a CRO working from an outdated protocol version.
- Defined Review Timelines: Contractually established service-level agreements (SLAs) for document review cycles ensure timely feedback and maintain study momentum.
- Clear Escalation Pathways: A pre-defined process for escalating issues to the appropriate decision-makers enables prompt resolution when disagreements or significant problems arise.
By implementing this operational structure, sponsors and CROs create a transparent and defensible record of their collaboration, which is essential during a regulatory audit. This elevates the relationship from a simple vendor transaction to a strategic partnership focused on mutual compliance. You can see how these ideas fit into the bigger picture by reading up on pharmaceutical quality management systems.
The flow below illustrates the progression of core documents throughout a trial, underscoring the need for narrative consistency from the protocol's initial design to the final study report.
The TMF as the Single Source of Truth
The Trial Master File (TMF), whether paper-based or electronic (eTMF), is the official and complete story of a clinical trial. It contains all essential documents that allow for the reconstruction and evaluation of the study's conduct and data quality, thereby demonstrating compliance with Good Clinical Practice (GCP).
To a regulator, the integrity of the TMF is a direct reflection of the quality of the trial itself. A poorly maintained TMF is a significant compliance risk, suggesting potential deficiencies in trial management.
Maintaining the TMF requires a contemporaneous filing approach, meaning documents are filed as they are finalized, not retrospectively.
Effective TMF management includes:
- Develop a TMF Plan: This document outlines the expected content, structure, and naming conventions, often referencing the TMF Reference Model for industry-standard organization.
- Assign TMF Ownership: Clearly define responsibilities for filing specific documents and for performing quality control checks.
- Conduct Regular QC Reviews: Perform periodic reviews of the TMF throughout the study to identify and correct any filing errors or omissions in a timely manner.
These structured workflows for document management, collaboration, and TMF maintenance are not administrative burdens. They are the essential mechanisms that enable regulatory affairs pharmaceutical professionals to demonstrate a consistent state of compliance, ensuring that all trial-related activities are defensible, transparent, and inspection-ready.
Modernizing Regulatory Submissions for Greater Efficiency
The field of regulatory affairs in the pharmaceutical industry is undergoing a significant digital transformation. Traditional processes, reliant on the manual creation and management of static documents like Word files and PDFs, are becoming increasingly inefficient in the face of growing data complexity.
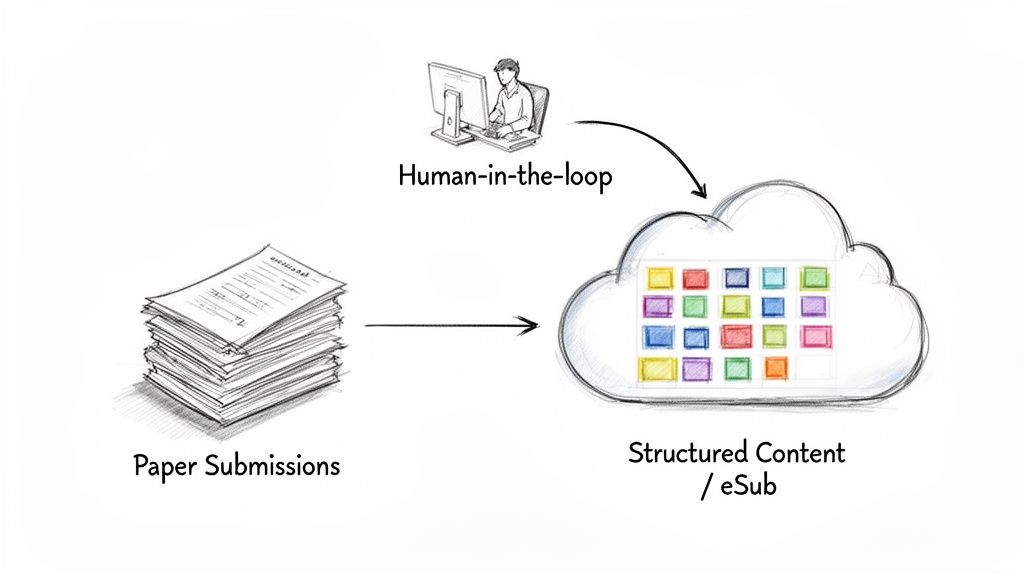
The modernization effort represents a paradigm shift from a document-centric to a data-centric approach. The objective is to manage critical information as structured, interconnected data components rather than as text locked within siloed documents. This evolution does not diminish the need for human expertise; instead, it allows regulatory professionals to redirect their focus from administrative tasks to strategic oversight and analysis.
From Disconnected Documents to Structured Content
In a traditional workflow, key information such as study objectives, endpoints, or safety parameters is often manually copied and pasted across numerous documents, including the protocol, investigator's brochure, and clinical study report. Any change to this information necessitates a labor-intensive manual update process across all relevant documents, introducing a high risk of inconsistency and error.
A modernized, structured content management approach deconstructs a document like a protocol into its fundamental components. The study title, objectives, and inclusion/exclusion criteria are managed as discrete, reusable content blocks.
This methodology establishes a single source of truth for core study information. When a component is updated, the change automatically propagates to every document that utilizes it, ensuring consistency and significantly reducing the need for manual quality control.
The operational impact of this shift is substantial. A McKinsey benchmark found that 80% of top pharma companies are re-engineering their Regulatory Information Management Systems (RIMS). This has resulted in submission timeline reductions of 50-65%, with some teams finalizing submissions within 8-12 weeks of database lock. For a product with $1 billion in peak sales, this acceleration can translate to an estimated $180 million in Net Present Value. You can get more details on how AI and zero-based design are reshaping submissions with McKinsey.
Operational Benefits of a Modernized Approach
Adopting a structured, data-centric workflow provides tangible benefits that extend beyond time savings to improvements in quality, risk management, and oversight. These advantages are realized through a combination of process control and technology, with human experts retaining control over critical decisions.
The table below contrasts the key characteristics of traditional and modernized regulatory workflows.
Traditional vs. Modernized Regulatory Workflow Comparison
| Aspect | Traditional Workflow | Modernized Workflow |
|---|---|---|
| Content Management | Relies on disconnected, static documents (e.g., Word files). | Manages content as structured, reusable data components. |
| Consistency | Maintained through manual checks and extensive QC cycles. | Enforced automatically through a single source of truth. |
| Change Management | Requires manual updates across dozens of documents, introducing high risk of error. | Changes propagate automatically to all linked documents. |
| Oversight | Limited to periodic reviews; difficult to track real-time progress. | Provides continuous, real-time visibility into document status and content. |
| Health Authority Queries | Higher risk of queries due to inconsistencies and formatting errors. | Lower risk of queries by ensuring data integrity and submission readiness. |
Ultimately, this modernization empowers regulatory teams to produce higher-quality submissions more efficiently. By automating repetitive, low-value tasks, it enables skilled professionals to concentrate on data interpretation, strategic narrative development, and proactive engagement with regulators.
Your Questions Answered: A Real-World Look at Regulatory Affairs
This section addresses common questions about the practical aspects of working in the field of pharmaceutical regulatory affairs, which combines science, law, and business strategy.
What's the One Skill Every Regulatory Affairs Pro Absolutely Needs?
The most critical skill is the ability to combine strategic thinking with meticulous attention to detail. A comprehensive knowledge of regulations is a prerequisite, but it is not sufficient on its own.
Effective regulatory professionals must be able to analyze clinical data from a regulator's perspective, anticipating potential questions and identifying compliance risks before they become issues. They must then construct a submission narrative that is both scientifically robust and fully compliant. This requires strong project management, clear communication, and the ability to align diverse functional teams—from clinical development to marketing—toward a common regulatory objective.
How Different Is Working in Big Pharma vs. a Small Biotech?
While the core function is the same, the operational environment differs significantly between large and small organizations.
- Big Pharma: Roles are often highly specialized within large, well-resourced teams. A professional might focus on a specific therapeutic area, a single stage of development (e.g., early-phase), or a particular geographic region. Systems and processes are well-established, but navigating the internal organizational structure can be complex.
- Small Biotech: Professionals typically have broader responsibilities. In a startup, an individual may constitute the entire regulatory department, handling everything from IND drafting to direct interactions with health authorities. This environment demands resourcefulness and adaptability but provides extensive exposure across the entire drug development lifecycle.
The primary distinction is between scope and depth. A large pharmaceutical company offers the opportunity to develop deep expertise in a specific area, while a small biotech provides a broad, end-to-end view of the regulatory landscape.
What's the Toughest Part About Managing Global Clinical Trial Submissions?
The most significant operational challenge is managing the variety of country-specific requirements while maintaining a cohesive global strategy. Although ICH guidelines aim to create a common framework, substantial differences in regulatory requirements persist between countries.
For instance, conducting a trial in 15 countries simultaneously requires managing unique submission formats, varying review timelines, and numerous local administrative requirements. One country may have specific requirements for the translation of patient-facing documents, while another may have a complex process for importing the investigational product.
Synchronizing these disparate activities without compromising the global project timeline is a major logistical undertaking. It demands meticulous planning and robust systems for tracking and managing every local variation.