A clinical trial protocol template serves as the standardized framework for developing a study's master plan. It is the foundational document that ensures all critical scientific, ethical, and operational details are structured in alignment with regulatory standards and Good Clinical Practice (GCP).
Understanding the Protocol as a Foundational Document
The clinical trial protocol is the single source of truth for a study, defining its objectives, methodology, and operational conduct. Its primary purpose is to safeguard trial participants and ensure the integrity of the collected data. A well-structured protocol is essential for clear communication among sponsors, CROs, clinical sites, and regulatory bodies.
A protocol evolves through a controlled process, transitioning from a basic structure to a final, approved document. This lifecycle management is critical for maintaining version control and ensuring the study team operates from the most current, authorized version.
The Evolution from Template to Final Submission
A common operational error is to treat the template as a simple fill-in-the-blanks document. In practice, the protocol has a distinct lifecycle, undergoing multiple rounds of review and revision before it is ready for regulatory submission or distribution to clinical sites.
This flowchart illustrates the typical progression of a protocol from a flexible template to a collaborative draft, and finally to the approved, version-controlled document.
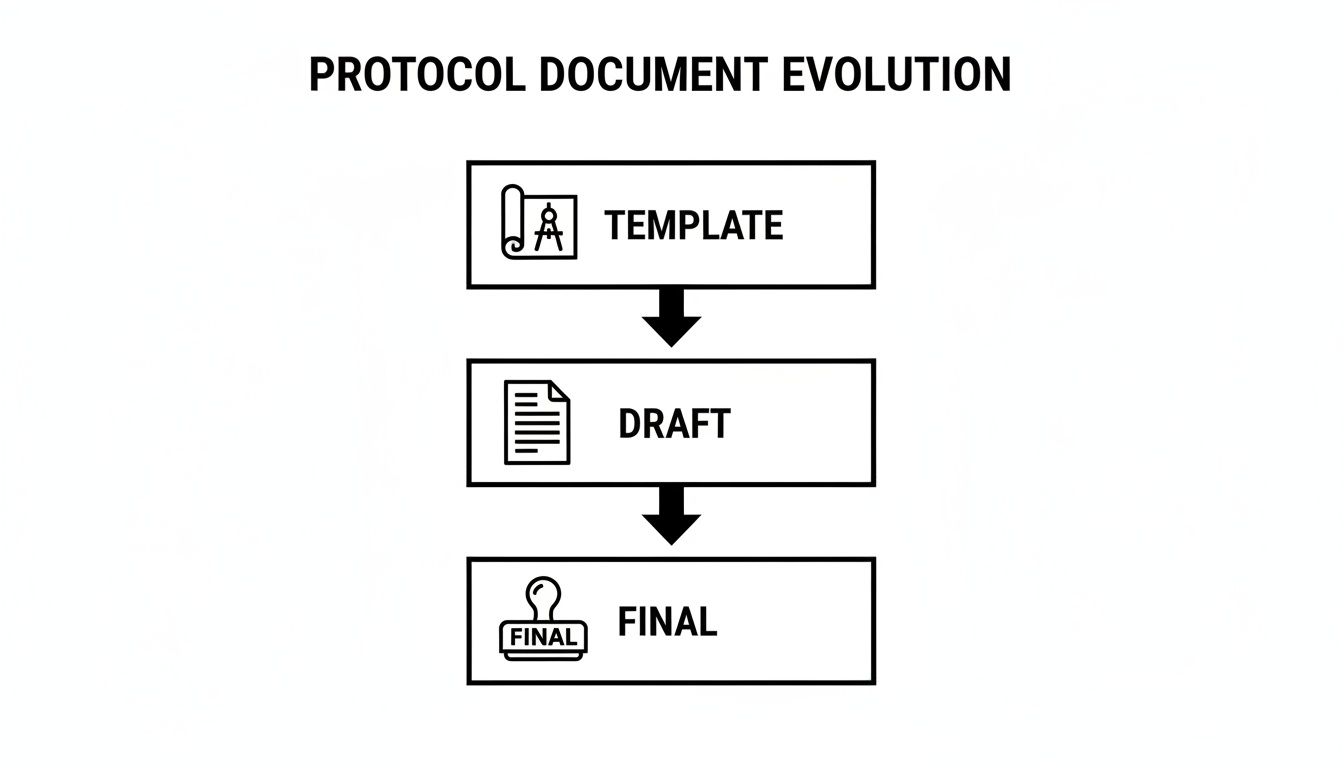
This progression distinguishes between the flexible starting point (the template), the collaborative work-in-progress (the draft), and the locked, final document that governs the trial.
Understanding these stages is fundamental for effective document lifecycle management, particularly within a controlled quality management system like Skaldi.
Core Components and Regulatory Alignment
The structure of a clinical trial protocol is dictated by global standards, primarily the International Council for Harmonisation (ICH) E6 guideline for Good Clinical Practice. ICH E6 provides a unified standard for the European Union, Japan, and the United States, detailing the essential content for a protocol. This standardization ensures that information is presented consistently, facilitating review by authorities such as the FDA and EMA.
The table below outlines the core sections required by ICH E6, which form the backbone of any compliant clinical trial protocol template.
Key Sections of an ICH E6 Compliant Protocol
| Protocol Section | Primary Purpose | Relevant ICH E6(R2) Section |
|---|---|---|
| General Information | Identifies the trial, sponsor, and key personnel. | 6.1 |
| Background Information | Provides the scientific rationale and summarizes relevant data. | 6.2 |
| Trial Objectives and Purpose | Defines the primary and secondary objectives of the study. | 6.3 |
| Trial Design | Describes the methodology, blinding, and randomization. | 6.4 |
| Selection of Subjects | Outlines inclusion, exclusion, and withdrawal criteria. | 6.5 |
| Statistical Methods | Details the plan for data analysis and sample size justification. | 6.9 |
Each section plays a specific role in creating a comprehensive plan for the clinical trial, ensuring regulatory requirements are addressed from the outset.
Enhancing Protocol Quality with SPIRIT and Global Standards
ICH E6 provides the regulatory foundation for a clinical trial protocol. For operational clarity and completeness, frameworks such as the SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) guidelines are widely used.
SPIRIT is not a regulatory requirement but an evidence-based set of recommendations designed to improve the quality and transparency of protocols. It provides a checklist of essential items, helping to prevent common omissions that often lead to protocol amendments. For multi-site trials, a SPIRIT-aligned template is a valuable tool for promoting consistency and clarity across all participating sites.
The Role of the SPIRIT Checklist
The SPIRIT checklist consists of 33 items recommended for inclusion in any protocol for an interventional trial. These items are grouped logically to align with standard protocol sections, covering administrative details, trial conduct, and data analysis. The objective is to ensure the protocol contains sufficient detail to allow for trial replication and critical appraisal by reviewers.
For example, while ICH requires a description of the trial design, SPIRIT prompts for more specific details on:
- Assignment of interventions: The methodology for randomization, sequence generation, and allocation concealment.
- Blinding: A precise description of who is blinded and the procedures used to maintain the blind.
- Data collection methods: Plans for data collection, management, and quality assurance for each study visit.
Incorporating these prompts into a template strengthens the document from its inception.
The Operational Impact of Adhering to SPIRIT
Integrating SPIRIT principles addresses documented deficiencies in protocol development. The SPIRIT 2013 guideline targeted critical areas where an estimated 70-80% of protocols were historically lacking key information. The global adoption of SPIRIT has been associated with a reduction in protocol amendment rates by 25-30%. Its influence is evident in the NIH's incorporation of its principles into their protocol templates to improve submission quality, a topic discussed in publications on the JAMA Network.
A well-designed template that aligns with SPIRIT functions as an embedded quality control check during the drafting process. It prompts the study team to address operational details early, reducing ambiguity that can lead to site queries, ethics committee questions, and regulatory delays.
This proactive approach helps produce a final protocol that is not only compliant but also operationally robust, facilitating a more efficient study startup and execution.
Drafting Key Administrative and Scientific Sections
The initial sections of a clinical trial protocol—including the Title Page, Protocol Summary, and Introduction—establish the study's identity, context, and scientific basis. They provide a concise overview for reviewers, such as regulators and Institutional Review Board (IRB) members.
Correctly drafting these foundational components is essential for creating a clear, compliant, and logical narrative. This is where the scientific rationale is presented, summarizing the nonclinical and clinical data that justify the trial in alignment with regulatory expectations from the FDA (21 CFR 312) and ICH guidelines.
Crafting the Title Page and Protocol Summary
The Title Page serves as the study's official identifier. Each detail is functional, required for regulatory tracking, site management, and operational control.
The Title Page must include these key elements:
- Full Protocol Title: A descriptive title stating the investigational product, phase, and target population.
- Protocol Identifying Number: The unique, sponsor-assigned number for version control.
- Sponsor and Investigator Information: Full names and addresses of the sponsor and coordinating investigator.
- Version and Date: The current version number and date to track amendments and prevent versioning errors.
The Protocol Summary (or Synopsis) follows the Title Page. For many IRB members and regulators, this is a frequently referenced section. It should be a self-contained summary of the full document.
A well-written summary can facilitate the review process. It should be concise, typically one to three pages, and logically present the study’s core elements: objectives, endpoints, design, and patient population.
Building the Scientific Rationale in the Introduction
The Introduction, or Background section, presents the scientific justification for the trial, as required by ICH E6 (Section 6.2). It should explain the existing knowledge and the critical research question the study aims to answer, thereby demonstrating that the trial is both necessary and ethically sound.
The narrative should include:
- The Investigational Product: A brief description of the product, its mechanism of action, and its class.
- Nonclinical Data: A summary of key findings from pharmacology, toxicology, and other preclinical studies that support testing in humans.
- Existing Clinical Data: A review of available human data from prior trial phases or related compounds.
- The Disease or Condition: An overview of the target condition, unmet medical needs, and the current standard of care.
Visualizing the Study with a Schema
A Study Schema, or flowchart, is a valuable tool for translating complex procedures and timelines into a simple diagram. For clinical site staff, this visual often serves as a quick reference for understanding a participant's journey through the trial.
An effective schema should visually map out:
- Study Periods: Clearly labeled phases such as screening, randomization, treatment, and follow-up.
- Visit Timelines: All key study visits and their timing.
- Treatment Arms: Distinctions between each cohort or treatment group.
- Major Procedures: High-level assessments or critical interventions planned at specific points.
Careful development of these initial sections creates an operationally clear and logical foundation, ensuring a shared understanding of the study's purpose and design among all stakeholders.
Study Objectives, Endpoints, and Design
After establishing the administrative details and scientific background, the protocol addresses the objectives, endpoints, and design. These sections form the scientific blueprint of the study. The objectives state what the study aims to discover, the endpoints define how this will be measured, and the design outlines the methodology to be used.
The precision of this framework is critical, as it dictates subsequent trial activities, from participant enrollment to data analysis.
Articulating Study Objectives
Study objectives are clear statements that explain the purpose of the trial. They should logically follow from the research question and be unambiguous.
Objectives are typically categorized by importance:
- Primary Objective: The single most important question the study is designed to answer. There should only be one. The trial's design, including the sample size calculation, is centered on addressing this question with sufficient statistical power.
- Secondary Objectives: Additional pre-planned questions that address other clinically relevant effects of the treatment.
- Exploratory Objectives: Objectives for generating new hypotheses to be tested in future studies. The trial may not be powered to provide definitive answers for these objectives.
Selecting and Defining Endpoints
Endpoints are the specific measurements used to assess the study objectives. Each objective must have at least one corresponding endpoint. The selection of endpoints is a critical decision that dictates the data to be collected and analyzed.
Endpoints are categorized in parallel with the objectives:
- Primary Endpoint: The single measurement that will address the primary objective. It must be clinically relevant, precisely defined, and reliably measured.
- Secondary Endpoints: Measurements that align with the secondary objectives, which can include additional efficacy metrics or safety outcomes.
- Exploratory Endpoints: Data points related to exploratory objectives, often used to identify trends or potential biomarkers.
According to ICH E9 Statistical Principles for Clinical Trials, the primary endpoint must be "the variable capable of providing the most clinically relevant and convincing evidence directly related to the primary objective of the trial."
For example, in an oncology trial with a primary objective to evaluate the effect of a new drug on tumor growth, the primary endpoint might be Progression-Free Survival (PFS). A secondary endpoint could be the Overall Response Rate (ORR), while an exploratory endpoint might be a change in a specific biomarker level.
Structuring the Study Design
The study design section describes the trial's architecture and methodology. It should provide a clear understanding of how the study will be conducted.
Key elements of the study design include:
- Description of the Trial: A high-level summary, for example: "This is a Phase 3, randomized, double-blind, placebo-controlled, parallel-group study."
- Randomization and Blinding: An explanation of how participants will be assigned to groups and what measures will be taken to minimize bias. The blinding method (e.g., double-blind, single-blind, open-label) should be specified.
- Control Group: A description of the comparator (e.g., placebo, active control, standard of care) and the rationale for its selection.
- Study Duration: The expected duration of participation for each subject and the overall trial timeline.
Participant Management and Data Analysis
This section of the protocol details the operational plans for managing participants and the data collected. These procedures are critical for protecting participant safety, ensuring data integrity, and defining the analytical methods for assessing study outcomes. Clear and detailed plans, aligned with global standards like ICH E6, are essential to mitigate trial risk.
Defining the Study Population and Assessments
Enrolling the appropriate patient population is fundamental to a clinical trial's validity. Inclusion and exclusion criteria serve two main purposes: to protect the safety of participants and to create a homogenous study population suitable for scientifically valid analysis.
- Inclusion Criteria are the characteristics a participant must have to be eligible for the study.
- Exclusion Criteria are the conditions that would disqualify a potential participant.
These criteria must be specific, objective, and rationally justified. For example, instead of a vague exclusion like "significant liver disease," a protocol should specify a quantitative measure, such as "Alanine aminotransferase (ALT) > 3 times the upper limit of normal." This precision ensures consistency across clinical sites.
The Study Assessments section outlines every procedure, test, and observation to be performed during the trial. This is typically presented in a Schedule of Assessments (SoA) table, which serves as a quick-reference guide for site staff, detailing the required activities for each study visit.
Example Schedule of Assessments (SoA)
The table below is a simplified example of a Schedule of Assessments, designed to provide site personnel with a clear overview of procedural requirements at each timepoint.
| Procedure/Assessment | Screening | Day 1 (Baseline) | Week 4 | Week 8 | End of Study |
|---|---|---|---|---|---|
| Informed Consent | X | ||||
| Physical Examination | X | X | X | X | X |
| Vital Signs | X | X | X | X | X |
| 12-Lead ECG | X | X | X | ||
| Blood Sample (PK) | X | X | X | ||
| Adverse Event Review | X | X | X | X |
This tabular format is critical for ensuring protocol adherence and consistency across all sites.
Summarizing Statistical Methods
While detailed statistical procedures are documented in a separate Statistical Analysis Plan (SAP), the protocol must provide a summary of the analytical approach, as described in ICH E9.
This summary should cover:
- Sample Size Justification: An explanation of how the number of participants was determined, including assumptions for statistical power, significance level, and expected effect size.
- Analysis Populations: Definitions of the different analysis sets, such as the Intent-to-Treat (ITT) and Per-Protocol (PP) populations.
- Statistical Models: A description of the primary statistical methods to be used for analyzing the primary and secondary endpoints.
This overview provides regulators and ethics committees with confidence that the trial is based on a sound statistical foundation.
Establishing Safety Reporting Procedures
Participant safety is paramount. This section of the protocol must define the procedures for identifying, documenting, and reporting adverse events (AEs). It must provide clear definitions and timelines that align with regulatory requirements from authorities like the FDA and EMA.
An Adverse Event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product, which does not necessarily have a causal relationship with this treatment. A Serious Adverse Event (SAE) is any AE that results in death, is life-threatening, requires inpatient hospitalization, or results in persistent or significant disability/incapacity.
The protocol must specify deadlines for investigators to report SAEs to the sponsor, for example, within 24 hours of awareness. This operational clarity is essential for maintaining regulatory compliance and vigilant safety oversight. More information on this topic can be found in our articles on clinical trial management.
How Standardized Templates Streamline Regulatory Submissions
Adopting a standardized clinical trial protocol template introduces measurable efficiencies that can positively impact trial timelines and budgets. A structured template can reduce authoring time, minimize clarification cycles with IRBs, and decrease the frequency of protocol amendments.
This approach brings predictability to the documentation process. When a protocol follows a consistent format, reviewers at regulatory agencies, ethics committees, and clinical sites can locate critical information more easily. This clarity can accelerate the study startup process. A robust structure ensures that all essential elements, such as safety monitoring plans and statistical methods, are included and adequately detailed from the outset.
Reducing Amendments
A significant benefit of a standardized template is the reduction of avoidable protocol amendments. Amendments are resource-intensive, introduce operational risk, and can cause study delays. A well-designed template functions as a quality control tool, prompting the study team to address key logistical details early in the development process.
This proactive approach reduces the ambiguity that often leads to clarification requests from sites or formal queries from an IRB. By embedding regulatory requirements and operational best practices into the template, teams are guided toward creating a complete, clear, and executable document from the start. A regulatory document management system is instrumental in managing such a controlled documentation process.
Evidence of Efficacy
The benefits of standardization are well-documented. For example, the NIH's implementation of standardized protocol templates has been shown to reduce preparation time by 30-40% and decrease the rate of protocol amendments from 35% to 15%. Prior to template adoption, IRB rejection rates were 50% higher due to incomplete Data Safety Monitoring Plans (DSMPs). Following adoption, overall approval rates increased by 20%.
These metrics indicate that a standardized clinical trial protocol template is a strategic asset for achieving operational excellence. It provides a foundation of clarity and completeness that can lead to more efficient regulatory submissions and faster trial initiation.
The Future of Protocol Development with ICH M11
The industry is moving toward a digital, harmonized, and machine-readable protocol format, guided by the International Council for Harmonisation's M11 guideline.
At the core of this initiative is the CeSHarP—the Clinical Electronic Structured Harmonised Protocol. This represents a new approach to creating and utilizing protocols. The objective is to establish a single, globally standardized format for protocol content and structure to improve the efficiency of data exchange between systems and with regulators.
How CeSHarP Functions
The primary innovation of ICH M11 is making the protocol machine-readable. A single, electronically structured format allows computer systems to interpret and act on protocol information, automating tasks such as populating clinical trial registries, configuring EDC systems, and streamlining regulatory review. This reduction in manual data entry can lead to fewer errors and shorter timelines.
CeSHarP is a structured data model, not merely a document. It deconstructs protocol information into specific data elements with controlled vocabularies, ensuring consistency across sponsors and regions.
The ICH estimates the CeSHarP template could reduce regulatory review times by up to 50% across the 15,000+ trials conducted annually in ICH regions.
Pilot programs have demonstrated 90% interoperability, addressing inconsistencies that previously caused 30-40% of regulatory questions in multi-regional trials.
This represents a fundamental shift from authoring narrative documents to creating structured, data-driven content. It requires medical writers and clinical operations teams to adopt a new mindset, functioning more like data architects.
As the industry adopts this new standard, understanding its principles will be essential for all professionals involved in protocol development.
Protocol Template FAQs
This section addresses common questions regarding the creation, use, and management of clinical trial protocol templates, with answers based on regulatory standards and operational best practices.
How Often Should a Protocol Template Be Updated?
A protocol template should undergo formal review at least annually. However, it should be updated immediately in response to significant regulatory changes, such as a major revision to ICH E6 or new guidance from the FDA.
The template should be managed as a controlled document within a Quality Management System (QMS). This ensures the template remains compliant with current regulations and reflects contemporary operational practices, preventing the use of outdated information in new protocols.
What is the Difference Between a Protocol and a SAP?
The protocol provides a high-level overview of the study's statistical approach, as required by ICH E6. It defines the primary and secondary endpoints and outlines the overall analysis strategy without detailing specific statistical methods.
The Statistical Analysis Plan (SAP) is a separate, detailed technical document. The SAP specifies the granular methodology for every planned analysis. It serves as the operational guide for biostatisticians and is typically finalized just before database lock. Its level of detail is too specific for inclusion in the main protocol.
Can One Template Work for All Clinical Trial Types?
A single template cannot adequately serve all clinical trial types. An ICH-based template is a solid foundation but requires adaptation for different study types, phases, and therapeutic areas. A master template should be viewed as a flexible framework rather than a rigid, one-size-fits-all document.
Specific adjustments are necessary for different trial types:
- Medical Device Trials: The protocol must include sections on device accountability and traceability, often referencing standards like ISO 14155.
- Adaptive Design Trials: These require a dedicated section outlining the adaptive methodology, pre-specified decision rules for modifications, and the unique statistical considerations of the design.
- Gene Therapy Studies: Protocols for these trials need expanded detail on long-term safety and efficacy follow-up to address specific regulatory expectations for advanced therapies.
Effective template management involves maintaining a core master template and creating specialized versions as needed. This approach balances consistency across a portfolio with the necessary customization for individual studies.