The European Union's Clinical Trials Regulation (CTR) represents a fundamental shift in the operational management of clinical trial documentation. The transition from separate, national submissions under the former Clinical Trials Directive (CTD) to a single, centralized system—the Clinical Trials Information System (CTIS)—necessitates significant adjustments to documentation workflows. The regulation's core tenets of harmonization and transparency require enhanced cross-functional alignment, standardized document structures, and proactive planning for public disclosure.
How the EU CTR Redefines Clinical Trial Documentation
The EU Clinical Trials Regulation (EU) No 536/2014 was designed to streamline the assessment and supervision of clinical trials across the European Union. It replaced the previous patchwork of national rules under the CTD with a unified regulatory framework.
The primary enabler of this framework is the Clinical Trials Information System (CTIS), a single online portal for all trial-related submissions, modifications, and communications between sponsors and regulatory authorities. For professionals managing clinical trial documentation, this shift from disparate systems to a central platform alters the entire lifecycle of document authoring, review, approval, and management. It places sponsors, CROs, and regulators within a common environment governed by legislated timelines, fundamentally reshaping operational processes.
A New Era of Harmonization and Transparency
The CTR is founded on two principles that directly influence documentation workflows:
- Harmonization: Sponsors now prepare a single, consolidated application dossier via CTIS, which is assessed jointly by all concerned Member States. This operational model demands robust internal alignment to ensure all documents are consistent and suitable for simultaneous multi-authority review.
- Transparency: A significant portion of the clinical trial dossier is made publicly available on the CTIS portal by default. Documentation workflows must now incorporate processes for identifying and redacting commercially confidential information (CCI) and personal data from the initial drafting stages, rather than as a final step before publication.
Since CTIS became mandatory for all new trial applications on January 31, 2023, operational challenges have been noted. Early user feedback highlighted workflow impediments such as document upload failures and difficulties with application updates, contributing to processing delays. The European Medicines Agency has shared more details on these initial system performance metrics.
The CTR transforms documentation from a series of siloed, country-specific tasks into a continuous, collaborative, and public-facing process. Operational readiness now depends on aligning internal workflows with the centralized structure and strict timelines of CTIS.
Ultimately, the regulation requires clinical documentation to be managed as a strategic operational function that demands meticulous planning and execution from the outset of a trial. To better understand the scale of this change, it is useful to compare the previous operational model under the CTD with the new reality under the CTR.
Key Documentation Workflow Shifts from CTD to CTR
| Workflow Aspect | Previous Process (Clinical Trials Directive) | Current Process (EU Clinical Trials Regulation) |
|---|---|---|
| Submission Model | Decentralized; separate submissions to each Member State's National Competent Authority (NCA) and Ethics Committee (EC). | Centralized; a single application dossier submitted via the Clinical Trials Information System (CTIS) for all concerned Member States. |
| Document Versioning | Multiple country-specific versions of documents (e.g., protocol, ICF) were common, leading to complex tracking. | A single "master" version for core documents is the standard. Country-specific adaptations are handled as annexes or within specific sections. |
| Review & Approval | Sequential or parallel reviews by different NCAs and ECs, each with their own timelines and feedback loops. | A coordinated assessment led by a Reporting Member State (RMS) with contributions from all concerned Member States, all within strict, legislated timelines. |
| Transparency | Limited and inconsistent public disclosure requirements, often occurring long after trial completion. | Proactive transparency is built-in. Documents are made public on CTIS by default, requiring upfront redaction of personal and commercially confidential data. |
| Amendments | Substantial amendments required separate submissions and approvals in each country, often a lengthy and disjointed process. | All substantial modifications are submitted centrally via CTIS and assessed simultaneously by all Member States, streamlining the update process. |
| Communication | Communication was fragmented, occurring directly and separately with each national authority via email, phone, or local portals. | All official communication, including Requests for Information (RFIs), is managed and documented within the CTIS portal. |
As the table illustrates, the CTR replaces the previous system with an integrated, highly visible workflow, shifting the focus from managing multiple independent processes to mastering a single, harmonized one.
Understanding the CTIS Centralized Dossier Structure
The Clinical Trials Information System (CTIS) serves as the central environment for sponsor-regulator interactions throughout the trial lifecycle. Mastering its dossier structure is essential for effective documentation workflows.
The CTIS application dossier is divided into two parts. This design separates the scientific basis of the trial from its country-specific implementation details, which in turn dictates how documents must be authored, reviewed, and managed.
Part I and Part II of the Dossier
This two-part structure organizes documents based on their scope and assessment pathway, directly influencing the eu clinical trials regulation (ctr) impact on documentation workflows.
-
Part I: Scientific Core Documents. This section contains multinational documents that undergo a single, joint assessment by all concerned Member States. It includes the protocol, the Investigator's Brochure (IB), and the Investigational Medicinal Product Dossier (IMPD). These documents must be harmonized and consistent across all participating countries.
-
Part II: National Implementation Documents. This section holds documents reviewed by each Member State individually. It includes recruitment arrangements, Informed Consent Forms (ICFs), and other subject-facing materials. These documents are based on the Part I foundation but are adapted to meet national requirements.
This structure requires seamless internal collaboration. A modification to a Part I document, for instance, may necessitate corresponding updates to Part II documents in multiple countries. Functional silos between medical writing, regulatory affairs, and clinical operations are operationally untenable in this model.
The system moves from fragmented, country-specific submissions toward a single, unified dossier where transparency is a core component.
Impact on Day-to-Day Workflows
The two-part dossier structure creates significant operational dependencies. The processes of medical writers, regulatory specialists, and clinical operations teams must be tightly integrated to maintain consistency and meet CTIS timelines.
For example, a Request for Information (RFI) from a regulator concerning a Part I document like the protocol could trigger required changes to Part II documents, such as the ICF, across multiple countries. The internal workflow must be capable of managing this cascade of updates efficiently and accurately.
The CTIS dossier necessitates a "single source of truth" model. The scientific core of the trial (Part I) must be finalized and internally agreed upon before country-specific documents (Part II) are completed. This requires disciplined version control and a high degree of cross-functional collaboration.
This principle aligns with the logical organization of a well-structured Trial Master File (TMF). Established frameworks like the DIA Trial Master File Reference Model provide a useful reference for organizing trial documentation effectively.
Maintaining Harmonization Across the Dossier
A critical operational challenge is ensuring consistency between Part I and Part II documents. The scientific details described in the protocol (Part I) must align perfectly with the information presented to a participant in an ICF (Part II). Any discrepancy can lead to regulatory queries and submission delays.
Internal review cycles must therefore include a quality control step where all Part II documents are systematically cross-referenced against the final, approved versions of Part I documents. This check should occur prior to submission, as all parties—sponsors, CROs, and regulators—view the same files within CTIS.
Implications of the CTR for Key Trial Documents
The EU Clinical Trials Regulation (CTR) extends beyond submission mechanics to reshape the content, structure, and lifecycle of critical clinical documents. The centralized assessment model means that documents such as the protocol, Investigator’s Brochure (IB), and Informed Consent Form (ICF) are reviewed as an interconnected package. This dynamic places a premium on inter-document consistency.
The Protocol and Substantial Modifications
Under the CTR, the protocol serves as the single source of truth for the trial across all participating Member States, eliminating country-specific variations of the core document. The most significant workflow change, however, relates to the management of amendments.
The previous process of submitting amendments on a country-by-country basis is replaced by a single substantial modification filed through CTIS. This action initiates a synchronized review by all concerned Member States, led by the Reporting Member State (RMS).
While this streamlined process improves efficiency, it concentrates pressure on internal operations. For example, the addition of a new secondary endpoint requires the preparation of a single modification package that is scientifically sound and operationally viable for all participating countries.
For any substantial modification, complete internal alignment must be achieved before submission. The package must anticipate and address potential concerns from all involved Member States, as the opportunity for negotiation is limited once the legislated review timeline begins.
Since CTIS became mandatory on January 31, 2023, the system has processed submissions from both commercial (approx. 60%) and non-commercial (approx. 40%) sponsors, the majority being multinational trials. This centralization has driven significant changes in document management. Timelines for substantial modifications, which previously could extend for up to 90 days per country, are now harmonized to a 60-day maximum. However, a European Commission survey indicated that many sponsors faced challenges in coordinating these modifications due to residual national-level procedural differences, leading to an increase in documentation revision cycles.
The Investigator’s Brochure and Safety Information
The Investigator's Brochure (IB) is also subject to procedural changes, particularly concerning the Reference Safety Information (RSI). The CTR encourages a synchronized update cycle for the IB and the Development Safety Update Report (DSUR).
An emerging best practice is to align the annual update of the IB’s RSI section with the data lock point for the DSUR. The updated IB is then submitted as part of a substantial modification package that includes the DSUR. This approach creates a predictable and transparent rhythm for managing safety information across the EU.
- Synchronized Timing: The IB update is linked to the DSUR reporting period.
- Harmonized RSI: The approved RSI becomes the definitive standard for assessing the expectedness of serious adverse reactions for the following year across all Member States.
- Streamlined Submission: The entire update is managed as a single, coordinated submission in CTIS.
This alignment simplifies safety reporting but requires meticulous planning. Teams must work in parallel to ensure the IB, DSUR, and any related protocol changes are prepared and reviewed concurrently.
Informed Consent Forms and Cross-Document Consistency
While the Informed Consent Form (ICF) remains a national-level document (Part II of the dossier), the CTR places it under greater scrutiny. Within CTIS, regulators can easily compare the protocol and ICF side-by-side, making inconsistencies immediately apparent.
Documentation workflows must now incorporate a dedicated cross-checking step. The information provided to participants in the ICF must accurately reflect the procedures, risks, and benefits detailed in the master protocol. Key areas for consistency checks include:
- Study objectives and procedures.
- Descriptions of risks and potential benefits.
- Participant rights and responsibilities.
Any divergence between the language in the ICF and the approved protocol is a significant compliance risk that can trigger regulatory queries and delay trial authorization. For further context on how these documents fit within the broader regulatory landscape, our overview of regulatory documents in clinical trials may be useful.
Clinical Study Reports and the Transparency Mandate
The CTR introduces a new, public-facing stage to the lifecycle of the Clinical Study Report (CSR). Transparency regulations require the submission of a summary of results to the public database within one year of the trial's conclusion. Subsequently, the full CSR is also published.
This mandate necessitates a "begin with the end in mind" approach to CSR authoring. Redaction of sensitive information can no longer be an afterthought. Workflows must include proactive steps to prepare a version suitable for public disclosure from the outset. This involves identifying and flagging commercially confidential information (CCI) and personal data during the drafting phase. Creating a "for publication" version of the CSR must become a standard component of the document finalization process.
Integrating Transparency into the Documentation Lifecycle
Under the EU Clinical Trials Regulation (CTR), transparency is an integral component of the documentation process. A large portion of the clinical trial dossier is required to be publicly available through the Clinical Trials Information System (CTIS) portal. This requirement fundamentally alters documentation workflows, demanding consideration of public disclosure from the initial drafting stage.
This represents a significant change from previous practices, where documents were prepared primarily for regulatory review, with public disclosure being a separate and later consideration. Now, key documents—from the protocol to the clinical study report—must be developed with a public audience in mind.
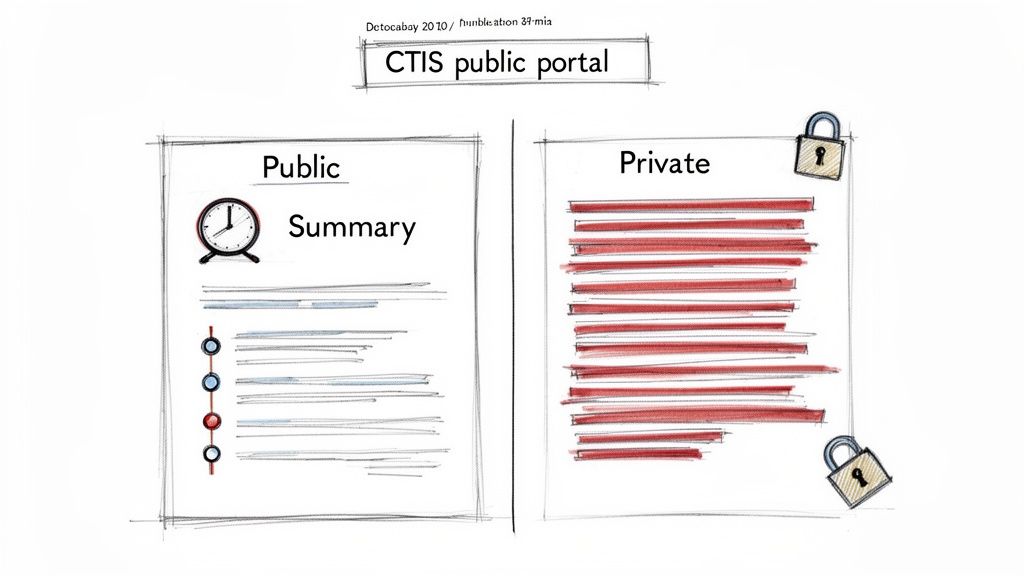
Establishing a Redaction Strategy Early
A primary operational challenge posed by the CTR's transparency rules is the need to redact commercially confidential information (CCI) and personal data. This cannot be a last-minute activity; it requires a systematic and defensible redaction workflow integrated into the entire process.
Organizations should first establish clear internal guidelines defining what constitutes CCI. These rules must be applied consistently to all documents intended for submission to CTIS. For example, specific manufacturing details in an IMPD or financial terms in contracts may be defined as CCI, but a robust justification must be prepared for each proposed redaction.
The CTR operates on a 'publish by default' principle. This places the burden of proof on the sponsor to justify why any information should be withheld from public view. A well-defined and consistently executed redaction strategy is essential for managing this requirement.
Proactive management is critical. Attempting to redact documents under tight submission deadlines increases the risk of compliance failures and the inadvertent disclosure of sensitive information. Redaction should be a formal, integrated step in the document review and approval cycle.
Building a ‘For Publication’ Document Workflow
To manage this requirement, SOPs should be adapted to accommodate two versions of key documents.
-
The Full, Unredacted Version: This is the complete document intended for Member State assessors, containing all scientific, operational, and confidential details required for their review.
-
The Redacted, ‘For Publication’ Version: This is the public-facing copy, prepared in parallel with the full version. It is scrubbed of all personal data and justified CCI and is the version that will be made available on CTIS.
This dual-version process should be formally integrated into the organization's documentation platform and workflows. A practical approach includes:
- Flagging at Source: Authors identify potential CCI during the drafting process.
- Cross-Functional Review: A designated team, typically including representatives from regulatory, legal, and clinical functions, reviews proposed redactions against the organization's CCI policy.
- Documenting Justifications: Each redaction must be logged with a clear, regulation-based rationale.
- Parallel Approval: Both the full and redacted versions should proceed through the final approval workflow simultaneously to ensure consistency and audit-readiness.
Managing Publication Timelines and Deferrals
The CTR specifies when different documents are made public. While some information is published shortly after a decision on the trial, sponsors can request to defer the publication of certain documents, such as the full protocol or Investigator’s Brochure.
A deferral is a delay, not an exemption. Workflows must account for these timelines by actively tracking each deferral period and ensuring the final, redacted "for publication" documents are ready for upload when the period expires. This requires robust project management and tracking capabilities within document management systems to ensure these critical deadlines are met. The objective is to make transparency a planned, predictable component of standard operations.
Realigning Team Collaboration Under the CTR
The EU Clinical Trials Regulation (CTR) necessitates a realignment of team collaboration. Sequential, siloed workflows are incompatible with the synchronized deadlines and unified dossier structure of the Clinical Trials Information System (CTIS). The regulation demands a shift toward parallel processing and continuous communication.
Medical writers, regulatory affairs specialists, and clinical operations staff must work in an integrated fashion to prepare and submit a single, harmonized dossier. The most significant eu clinical trials regulation (ctr) impact on documentation workflows is this requirement for a new, highly collaborative operational model.
Defining CTIS User Roles and Responsibilities
A foundational step is to define user roles and responsibilities within CTIS. The system features specific roles and permissions that must be mapped to the team's operational responsibilities to prevent process gaps and missed deadlines.
A responsibility matrix is an effective tool for this purpose:
- CTIS Administrators: A limited group responsible for user access management and overall system setup.
- Submission Preparers: Individuals responsible for uploading documents and assembling the dossier in CTIS, typically from medical writing and regulatory operations teams.
- Submitters: A tightly controlled role assigned to senior regulatory affairs professionals with the authority to submit applications and responses.
- Viewers: A broader, read-only role for team members who need to monitor a trial's progress without editing or submission privileges.
Every action within CTIS is logged and time-stamped, creating a permanent audit trail. Defining these roles is not only an efficiency measure but also a critical component of risk management, ensuring a clear record of who performed which actions and when.
Establishing a Rapid Response Process for RFIs
Requests for Information (RFIs) from regulators are a common occurrence in CTIS and come with strict, non-negotiable deadlines, often only 10-12 days. An inability to respond comprehensively within this timeframe can halt the trial application process.
Organizations must establish a rapid-response workflow that is documented, tested, and ready for immediate activation. A robust RFI process typically includes:
- Immediate Triage: A designated point of contact, often in the regulatory function, receives the RFI notification and immediately routes the query to the appropriate subject matter experts.
- Collaborative Drafting: Medical writers, clinicians, and statisticians work in parallel to draft the response and update any affected documents.
- Expedited Review: A streamlined internal review and approval process ensures the response is verified and signed off by key stakeholders well before the CTIS deadline.
The team's ability to execute this process efficiently is critical to maintaining momentum in the clinical trial application process.
Building a CTR-Ready Documentation Framework
Adapting to the EU Clinical Trials Regulation (CTR) requires more than minor SOP adjustments; it necessitates a comprehensive overhaul of documentation handling. The objective is to create a robust system that supports the regulation's principles of harmonization, transparency, and efficiency. This involves a critical assessment of current processes and tools to ensure they can meet the demands of the Clinical Trials Information System (CTIS).
A CTR-ready framework is built on proactive controls integrated into every stage of the document lifecycle, from drafting to archiving. It must be designed to accommodate parallel review cycles, synchronized timelines, and public disclosure requirements.
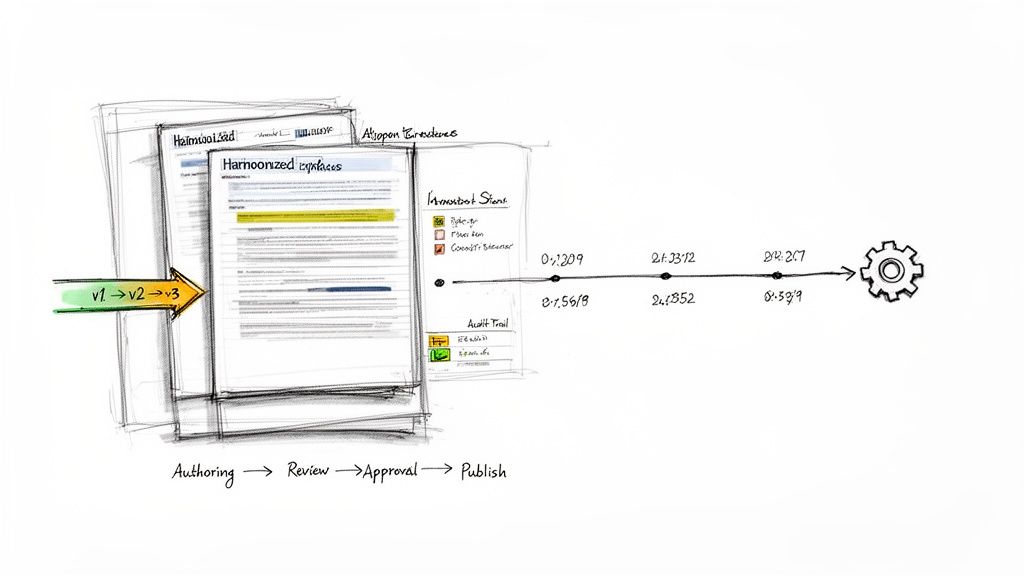
Implementing Structured and Harmonized Templates
A foundational element is the establishment of a single source of truth through structured, harmonized templates for core documents like protocols and Investigator's Brochures. The eu clinical trials regulation (ctr) impact on documentation workflows is most acute when teams attempt to use inconsistent or country-specific templates within a unified system, creating friction and rework.
Harmonized templates drive consistency from the outset. By standardizing sections and core content, they reduce variability between documents and streamline the review process. This ensures documents align with internal standards and CTR expectations before they are submitted to CTIS.
Enhancing Version Control and Audit Trails
The CTR's coordinated assessments create a significant operational challenge in managing concurrent reviews and rapid updates. A robust version control system is therefore essential. It must provide clear differentiation between draft, review, and final approved versions, which is particularly critical when managing parallel modifications to Part I and Part II documents.
Equally important is a comprehensive audit trail. Regulators expect a complete history of every document, including all reviews, comments, and signatures.
A CTR-ready framework ensures every action performed on a document is automatically logged. This creates a detailed, inspection-ready history that demonstrates control and compliance throughout the document's lifecycle—from initial draft to final submission and public release.
This level of granular tracking is fundamental for providing evidence of procedural adherence during a regulatory inspection.
Adopting a Controlled Documentation Platform
These components—structured templates, version control, and audit trails—are best managed within a controlled environment designed for regulatory content. Such platforms are engineered to support the compliant, human-in-the-loop workflows required for preparing high-stakes clinical documentation. They provide the necessary structure to manage templates, enforce version control, and maintain comprehensive audit trails within a single system.
For teams evaluating their current systems, understanding the core functions of a modern regulatory document management system is a logical starting point. Building this operational backbone creates a framework that not only addresses CTR compliance but also establishes long-term efficiency and inspection readiness.
Frequently Asked Questions
Common questions from clinical trial teams adapting their documentation practices for the EU CTR.
What is the primary documentation challenge when transitioning an ongoing study from the CTD to the CTR?
The most significant challenge is compiling a complete, CTR-compliant dossier for a study not originally designed for submission via CTIS. This requires a retrospective effort to ensure all existing documents—including the protocol and Investigator's Brochure—meet the new harmonized standards. This process often involves a comprehensive documentation audit and remediation to align legacy materials with the structured requirements of CTIS before the transition deadline.
How does the CTR change the process for managing substantial modifications?
The process for substantial modifications under the CTR is a prime example of its dual impact. On one hand, efficiency is gained by submitting a single modification via CTIS for a coordinated assessment by all concerned Member States. On the other hand, this centralizes pressure on internal coordination.
The entire documentation workflow must be oriented toward preparing a single, unified modification package that is acceptable to all participating countries upon initial submission. Any divergent feedback must be reconciled and addressed within a single, strict timeframe, making the quality of the initial package critical.
While the process eliminates redundant country-by-country submissions, the requirements for the unified submission are substantially higher.
How do the CTR’s transparency rules affect the authoring of a Clinical Study Report?
The transparency requirements fundamentally alter the CSR authoring process. The CTR mandates that a summary of the Clinical Study Report (CSR), and subsequently the full report, be made public. Consequently, the CSR can no longer be written solely for an internal or regulatory audience; public disclosure must be considered from the initial draft.
In practical terms, workflows must incorporate specific steps to address this:
- Create a lay-person summary in parallel with the technical report.
- Proactively develop a "for publication" version of the final CSR as an integral part of the finalization process.
- Systematically identify and redact commercially confidential information (CCI) and all personal data as part of the authoring and review cycle, rather than as a final, pre-publication step.