The ICH guidelines for clinical trials represent a harmonized international standard for the design, conduct, monitoring, recording, analysis, and reporting of clinical research involving human participants. This unified framework is intended to ensure that clinical data submitted to regulatory authorities, such as the FDA, EMA, or PMDA, is mutually acceptable across these regions.
The Purpose of ICH: Harmonisation for Global Trials
Before the establishment of the International Council for Harmonisation (ICH), sponsors often faced disparate regulatory requirements across different countries. A clinical trial conducted according to one country's standards might require significant modification or repetition to meet the demands of another, leading to inefficiencies and delays in drug development.
The ICH was formed to harmonize the technical requirements for registering new medicinal products. By creating a common framework for clinical research, the ICH facilitates a more streamlined development process for sponsors, contract research organizations (CROs), and regulators.
What Global Harmonisation Aims to Achieve
The core objective of the ICH is to promote public health by enabling the efficient development and registration of safe, effective, and high-quality medicines. This mission is supported by several key goals:
- Protecting Trial Participants: The guidelines establish standards to protect the safety, well-being, and rights of individuals participating in clinical trials.
- Minimizing Redundant Testing: By creating a single standard, harmonized guidelines reduce the need to conduct duplicative testing to meet different regional requirements, conserving resources.
- Improving Regulatory Review Efficiency: Submissions based on globally accepted standards for data collection and reporting can be reviewed more efficiently by regulatory agencies.
This framework enables multinational clinical trials, allowing data from a trial conducted in one ICH region to be accepted in others, a foundational element of modern drug development.
Foundational Guidelines for Clinical Trials
While the ICH has published numerous guidelines, a core set forms the basis for clinical trial operations and documentation. This guide will focus on these critical guidelines and their interrelationships.
ICH guidelines are not static regulations but an evolving framework designed to adapt to scientific and technological advancements. Their purpose is to ensure trial integrity and participant safety while accommodating innovative research methodologies.
This structured approach means that from the initial draft of a study protocol to the final clinical study report, each step is guided by principles of quality, ethics, and science. Understanding how these key documents interact is essential for preparing a successful regulatory submission.
ICH E6 Good Clinical Practice: The Cornerstone of Trial Conduct
ICH E6 Good Clinical Practice (GCP) serves as the operational standard for designing, conducting, monitoring, recording, analyzing, and reporting clinical trials. Its primary purpose is to protect the rights and safety of trial participants and to ensure the credibility and integrity of the data generated.
This guideline provides a common language and set of expectations for sponsors, investigators, and regulators. It establishes a global standard for ethical and scientific quality, ensuring consistency in trial conduct regardless of location.
From E6(R2) to E6(R3): An Evolution in Approach
For years, ICH E6(R2) was the established standard. However, the increasing complexity of trial designs, the rise of decentralized studies, and the integration of digital health technologies necessitated an updated framework.
The forthcoming ICH E6(R3) represents a significant revision of GCP standards, addressing the realities of modern trials that incorporate tools like eConsent and electronic health records (EHRs). It encourages a shift toward a quality by design (QbD) mindset.
By prospectively defining factors critical to quality, teams may be able to reduce documentation review cycles by implementing structured workflows compliant with E6(R3), E3, and E9. For further information, expert insights on the adoption of ICH E6(R3) are available.
The core principle behind E6(R3) is a move from a prescriptive, one-size-fits-all approach to a more flexible, risk-based framework. This allows trial teams to focus resources on activities that are most critical to participant safety and data integrity.
To illustrate the change, the following table compares the two versions.
Key Evolutions from ICH E6(R2) to E6(R3)
| Aspect | ICH E6(R2) Focus | ICH E6(R3) Emphasis |
|---|---|---|
| Overall Approach | A more prescriptive, one-size-fits-all methodology. | A flexible, risk-proportionate, "fit-for-purpose" framework. |
| Quality Management | Quality control and assurance as separate, often reactive, functions. | Proactive "quality by design" (QbD) integrated from the start. |
| Technology | Limited guidance on digital systems; paper-based mindset. | Explicitly embraces digital health technologies, EHRs, and e-systems. |
| Data Integrity | Focused on source data verification (SDV) as a primary tool. | Focuses on identifying and managing data critical to trial quality (CTQ factors). |
| Sponsor Oversight | Detailed requirements for on-site monitoring. | Encourages centralized and risk-based monitoring strategies. |
| Protocol Design | Traditional protocol structures assumed. | Supports innovative and complex trial designs (e.g., adaptive, decentralized). |
This table highlights the fundamental shift from a process-driven checklist to a principles-driven, agile approach better suited for the complexity of modern clinical research.
The Core Principles of GCP in Practice
ICH E6 is guided by a set of foundational principles that translate directly into how key documents are developed, managed, and executed.
- Ethical Conduct: Trials must adhere to the ethical principles originating in the Declaration of Helsinki.
- Benefit-Risk Assessment: The foreseeable benefits of a trial must outweigh the potential risks before initiation and throughout the study.
- Participant Rights and Safety: The rights, safety, and well-being of trial subjects take precedence over the interests of science and society.
- Adequate Product Information: Sufficient nonclinical and clinical data on the investigational product is required to support the proposed trial.
- Scientifically Sound Protocol: Trials must be scientifically sound and described in a clear, detailed protocol.
- IRB/IEC Approval: The protocol and related materials must be approved by an Institutional Review Board (IRB) or Independent Ethics Committee (IEC) prior to trial initiation.
- Qualified Personnel: Medical decisions must be made by a qualified physician or, where appropriate, a dentist.
- Data Quality and Integrity: Data must be recorded, handled, and stored in a way that allows for accurate reporting, interpretation, and verification.
Defining Key Roles and Responsibilities
A key function of ICH E6 is to clearly define roles and responsibilities, which is fundamental to oversight and accountability.
The Sponsor
The sponsor is the entity that initiates, manages, and finances the trial, bearing ultimate responsibility for its quality and integrity.
- Implement and maintain quality assurance and quality control systems.
- Select qualified investigators and provide them with necessary information, including the Investigator's Brochure (IB).
- Ensure proper monitoring and auditing of the trial.
- Report safety information to regulatory authorities and investigators as required.
The Investigator
The investigator is responsible for the conduct of the trial at a specific site.
- Ensure the trial is conducted in compliance with the approved protocol.
- Protect the rights, safety, and welfare of trial participants.
- Obtain valid informed consent from each participant before any trial-related procedures.
- Maintain accurate and complete source documents and trial records.
The Institutional Review Board / Independent Ethics Committee (IRB/IEC)
The IRB/IEC is an independent body responsible for protecting the welfare of human subjects.
- Review and approve the trial protocol, the informed consent form (ICF), and other participant-facing materials.
- Conduct continuing review of the trial to ensure ongoing participant safety.
This division of responsibilities creates multiple layers of oversight to uphold GCP principles and produce credible data for regulatory evaluation.
ICH E8: The Strategic Blueprint for Study Design
While ICH E6 provides the operational instructions for how to conduct a clinical trial, ICH E8 (R1) offers the strategic framework for why the study is designed in a particular way. Titled General Considerations for Clinical Studies, this guideline focuses on the overall clinical development lifecycle and encourages proactive, intentional design from the outset.
The core concept promoted by ICH E8 is Quality by Design (QbD). Instead of reacting to issues as they arise, QbD requires the prospective identification of factors that are critical-to-quality (CTQ)—elements essential for protecting participants and generating reliable data to answer the study's primary questions.
Defining Critical-to-Quality Factors
ICH E8 emphasizes addressing fundamental questions before the protocol is drafted to avoid ambiguity and inefficiency.
- Study Objectives: What is the precise scientific question the study aims to answer?
- Study Population: Who are the intended participants, and how do their characteristics inform the trial design and ethical considerations?
- Study Design: What trial design (e.g., placebo-controlled, active comparator, adaptive) is most appropriate to meet the stated objectives?
- Data Collection: What specific data are necessary to support the defined endpoints, avoiding the collection of extraneous information that adds complexity and risk?
By promoting this upfront planning, ICH E8 helps ensure that other guidelines, such as E6 for GCP and E9 for statistics, are applied to a well-reasoned and scientifically sound foundation. This can help prevent late-stage protocol amendments and operational challenges.
ICH E8 advocates for a cross-functional, collaborative approach to study design. It underscores that quality is a shared responsibility among clinicians, statisticians, data managers, and operations teams from the beginning of the development process.
From Strategy to Protocol Development
The principles in ICH E8 transform a protocol from a set of instructions into a strategic document. A protocol developed with an E8 foundation not only specifies what to do but also justifies why each aspect of the study design is necessary.
For example, when defining study endpoints, E8 encourages teams to select outcomes that are clinically meaningful to patients and prescribers, ensuring the trial produces results that are both statistically significant and relevant.
This strategic approach also impacts operational feasibility. By identifying CTQ factors early, data collection and monitoring plans can be designed to be proportionate to the identified risks. This aligns with the risk-based principles central to ICH E6(R3) and allows for a more targeted allocation of resources. Ultimately, ICH E8 aims to establish a strong foundation for a successful trial before it begins.
ICH E9: The Statistical Blueprint for Clinical Trials
While ICH E8 addresses the strategic "why" and ICH E6 the operational "how," ICH E9 provides the scientific framework for ensuring trial results are robust and unbiased. This guideline, Statistical Principles for Clinical Trials, outlines the statistical methods for designing, conducting, and analyzing trials to minimize bias and support credible conclusions.
ICH E9 emphasizes that statistical considerations must be integrated into the trial from the initial design phase, not applied retroactively. This proactive approach helps ensure the study is appropriately powered, uses suitable randomization methods, and follows a pre-specified plan for data analysis.
The E9(R1) Addendum and the Estimand Framework
The ICH E9(R1) addendum introduced the estimand framework, a structured approach to precisely defining the treatment effect to be measured. The estimand clarifies the specific scientific question the trial aims to answer, ensuring alignment between the study's objective, design, data collection, and analysis.
The estimand framework requires defining five components:
- Treatment: The investigational product and its conditions of use.
- Population: The specific patient population under study.
- Variable (Endpoint): The outcome measured to assess the treatment effect.
- Intercurrent Events: How events that occur after treatment initiation (e.g., use of rescue medication, discontinuation) will be addressed in the analysis.
- Population-Level Summary: The metric used to summarize the effect across the population (e.g., difference in means, ratio of proportions).
Defining these attributes upfront provides an unambiguous target for the study, which is critical for interpreting results and communicating them to regulators and clinicians.
The estimand framework from the ICH E9(R1) addendum reduces ambiguity in trial reporting. It ensures that when a "treatment effect" is reported, its meaning is clear to all stakeholders, including how intercurrent events like patient dropouts were handled.
Operationalizing Statistical Principles: The Statistical Analysis Plan (SAP)
The principles of ICH E9 are operationalized in the Statistical Analysis Plan (SAP). This detailed, technical document specifies, in advance, how the trial data will be analyzed. The SAP is typically finalized after the protocol is approved but before the data are unblinded.
A well-defined, E9-compliant SAP serves as a pre-specified guide for analysis, preventing data-driven analysis choices that could introduce bias (e.g., "p-hacking"). By pre-specifying all analyses, the SAP protects the scientific integrity of the findings. It defines the primary and secondary endpoints, methods for handling missing data, and the statistical models to be used. A comprehensive SAP is a cornerstone of a regulatory submission, providing reviewers with the transparency needed to evaluate the study's conclusions.
For more information on developing this document, a statistical analysis plan template can provide a useful starting point.
ICH E3: The Blueprint for Your Clinical Study Report
After a clinical trial is completed and the data have been analyzed, the findings are documented in the Clinical Study Report (CSR), a comprehensive and integrated account of the trial’s conduct and results. The ICH E3 guideline provides the globally recognized structure and content for this critical document, ensuring clarity, completeness, and consistency.
The CSR is a core component of a regulatory submission, such as a New Drug Application (NDA) or Marketing Authorisation Application (MAA). Its purpose is to present a single, cohesive narrative that allows regulators to understand the study's conduct, assess the validity of its findings, and, if necessary, re-analyze the data. ICH E3 provides the standardized organization that makes this extensive document navigable for reviewers worldwide.
The Mandated Structure of an E3-Compliant CSR
ICH E3 specifies a detailed table of contents that all compliant CSRs must follow. This standardized format is crucial for an efficient regulatory review, as it allows a reviewer in any ICH region to locate specific information quickly.
The structure progresses logically from a high-level overview to granular details. Key sections include:
- Title Page: Basic study identification information.
- Synopsis: A brief (typically ≤3 pages), standalone summary of the trial's design, conduct, and key findings.
- Table of Contents: A roadmap for the report and its appendices.
- List of Abbreviations and Definitions: A glossary of terms.
- Ethics: A statement confirming the trial was conducted in accordance with ethical principles.
- Investigators and Study Administrative Structure: A list of individuals and bodies involved in conducting and managing the trial.
- Introduction: The scientific rationale for the study.
- Study Objectives: A clear statement of the primary and secondary goals.
- Investigational Plan: A detailed description of the study design, patient population, treatments, and efficacy and safety measurements, which must align with the final protocol.
- Study Patients: Details on patient disposition, demographics, and baseline characteristics.
- Efficacy Evaluation: Presentation of the analysis of primary and secondary endpoints, as pre-specified in the SAP.
- Safety Evaluation: A thorough report on adverse events, laboratory data, and other safety measures.
- Discussion and Overall Conclusions: Interpretation of findings, a benefit-risk assessment, and final conclusions.
- Appendices: Includes source documents such as the protocol, sample case report forms, and patient data listings.
This prescribed format has been instrumental in standardizing regulatory dossiers, contributing to more efficient review times and improved data traceability. The official ICH guidance documents from the FDA provide further details.
Ensuring Data Traceability
An effective CSR does more than present data; it establishes a clear and unbroken line of traceability from the study's foundational documents to the final results. The narrative must demonstrate a consistent path from the objectives in the protocol, through the analysis methods in the SAP, to the results presented in the CSR.
The guiding principle of an ICH E3-compliant CSR is transparency. A regulator should be able to review the report and understand not only what happened in the trial but why it was conducted that way, without requiring additional clarification.
This traceability is a critical requirement. Any deviation from the protocol or SAP must be documented and justified within the CSR. For example, if a statistical method was altered from what was pre-specified, the rationale for the change and its potential impact on the results must be discussed. This builds confidence with regulators and supports a smoother review process. To facilitate consistency, starting with a well-structured document is beneficial. A guide to the clinical study report template can offer practical insights.
Integrating ICH Guidelines into Documentation Workflows
ICH guidelines should be viewed not as a final compliance check, but as an integral part of the entire clinical trial process. Adherence is demonstrated through a clear, logical, and traceable narrative woven into every document, from study concept to final submission.
The process begins with ICH E8, which establishes the strategic blueprint. The principles of Quality by Design (QbD) from E8 inform the protocol, ensuring that study objectives and critical-to-quality factors are defined at the outset. This protocol then governs the trial's operational conduct under ICH E6 (Good Clinical Practice), which includes the management of key documents like the Investigator’s Brochure and informed consent forms.
Mapping Guidelines to Key Documents
Concurrently, the principles of statistical integrity from ICH E9 are translated into the Statistical Analysis Plan (SAP). The SAP provides the technical instructions for analyzing the data collected under the GCP standards of E6, bridging trial conduct with data analysis.
Finally, the protocol's strategic plan, the trial's operational execution, and the SAP's analytical methods converge in the Clinical Study Report (CSR). The CSR, structured according to ICH E3, presents the definitive and comprehensive account of the trial.
The objective of an integrated workflow is to maintain a "golden thread" of logic and traceability. A regulator should be able to trace a study objective from the protocol through the methodology and results to its final conclusion in the CSR without inconsistencies.
This diagram illustrates how the Clinical Study Report (CSR) serves as a central document, integrating the protocol and SAP for submission to regulatory authorities.
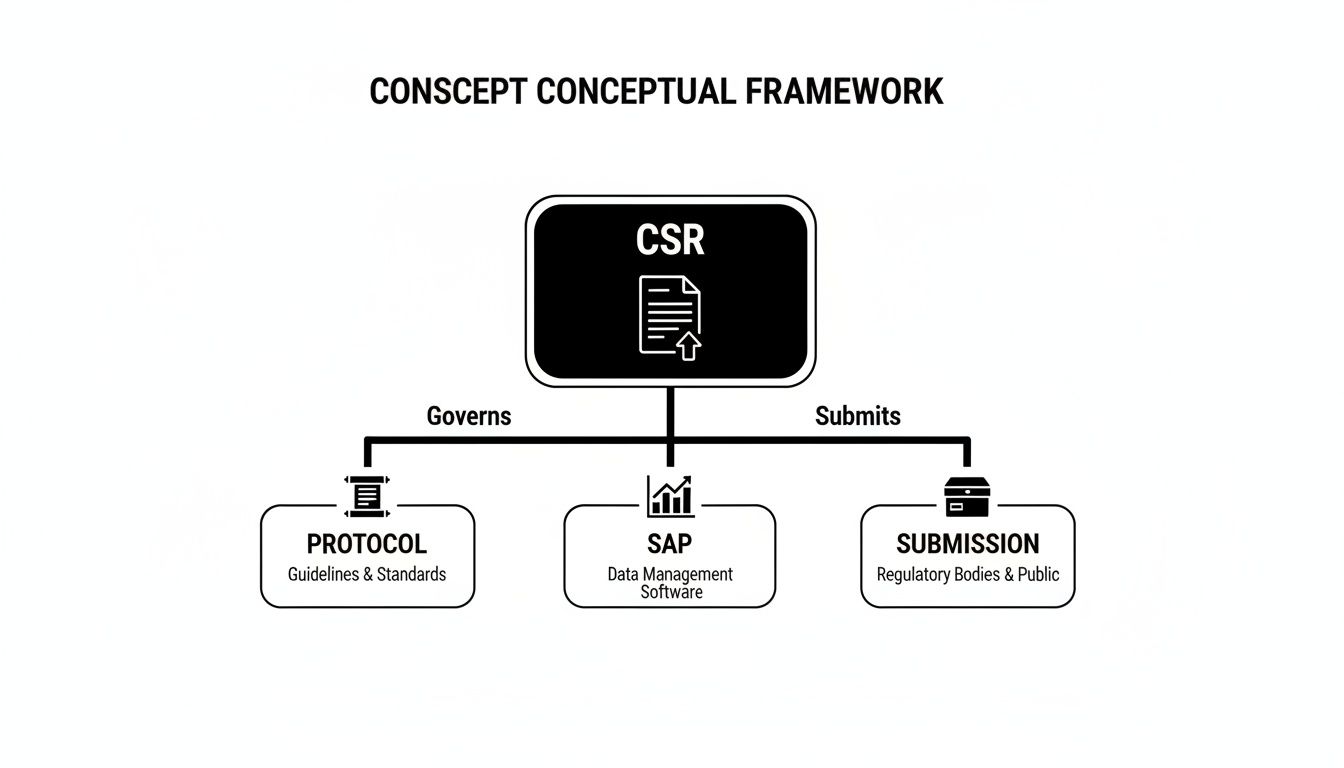
The CSR is the culmination of a well-defined process, demonstrating how these individual components must align to form a submission-ready package.
Achieving Consistency and Audit Readiness
For sponsors and CROs, this integrated approach is necessary to maintain consistency and audit readiness. Any protocol amendment must be reflected in all related documents. Deviations from the plan must be documented and justified to maintain the integrity of the study narrative.
Managing this level of consistency across numerous complex documents manually is challenging and introduces risk. A regulatory document management system can help manage this complexity. A structured, systems-based approach helps ensure that compliance is a continuous, manageable part of clinical trial execution rather than a final-stage remediation effort.
A Few Common Questions About ICH Guidelines
Professionals in clinical operations and regulatory affairs often have recurring questions about the practical application of ICH guidelines. This section addresses some of the most common inquiries.
How Do ICH E6, E8, and E9 Actually Work Together?
These three guidelines function as an interconnected framework, where strategy informs conduct and statistical principles provide validation.
-
ICH E8 (General Considerations for Clinical Studies): This is the strategic foundation. It promotes a Quality by Design (QbD) approach to define the "why" of the study and identify what is critical to quality before the trial begins.
-
ICH E6 (Good Clinical Practice): This is the operational standard—the "how." It provides the rules for the day-to-day conduct of the trial, ensuring ethical execution, participant safety, and data reliability.
-
ICH E9 (Statistical Principles for Clinical Trials): This is the scientific validation. It ensures the "what"—the trial's design and analysis—is statistically sound and free from bias, supporting credible results.
In short, E8 helps design the right trial, E6 ensures the trial is run right, and E9 provides confidence that the conclusions are right.
Are ICH Guidelines Actual Laws?
This is a key distinction. ICH guidelines are not laws in themselves; they are international recommendations developed to harmonize technical standards.
However, regulatory authorities in ICH regions, such as the FDA in the United States and the EMA in Europe, have adopted these guidelines as official guidance for the industry.
Therefore, while they are not statutes, they function as the effective standard for regulatory compliance. Failure to adhere to these guidelines will likely result in significant challenges during regulatory review, including delays, extensive queries, or rejection of a marketing application. For all practical purposes, they should be treated as mandatory.
What’s the Single Biggest Change in ICH E6(R3)?
The most significant shift in the upcoming ICH E6(R3) is the move from a prescriptive, one-size-fits-all GCP model to a flexible, risk-proportionate, and Quality by Design (QbD) framework. This change affects both documentation and operational practices.
For documentation, it means the protocol must clearly justify the choice of trial design, data collection methods, and monitoring strategies based on a formal risk assessment.
Operationally, the most notable impact is on monitoring. The previous convention of 100% source data verification (SDV) is being replaced by a risk-based approach. E6(R3) encourages focusing monitoring efforts on the data and processes most critical to participant safety and the achievement of the trial's primary objectives.